ARMIDA ZÚÑIGA ESTRADA, Comisionada Federal para la Protección contra Riesgos Sanitarios y Presidenta del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, con fundamento en lo dispuesto por el artículo 39 de la Ley Orgánica de la Administración Pública Federal; 4 de la Ley Federal de Procedimiento Administrativo; 3o, fracciones XVI y XXVI, 13, apartado A, fracción I, 17 Bis, fracciones III y VIII, 45, 313, fracciones I y III, 314 fracciones III, IV, VI y XIII, 315, 316, 317, 319, 321, 322, 323, fracción II, 325, 327, 340, 341, 341 Bis, 342 y 375 fracción VI, 459, 460, 461 y 462, fracción II de la Ley General de Salud; 10, fracción I, 30, 34, 35, fracción V, 37 y 38 de la Ley de Infraestructura de la Calidad; 28 y 33 del Reglamento de la Ley Federal sobre Metrología y Normalización en relación con el Transitorio Tercero del Decreto por el que se expide la Ley de Infraestructura de la Calidad y se abroga la Ley Federal sobre Metrología y Normalización; 4o, 38, 39, 40, 41, 42, 43, 44, 45, 46, 48, 53 y 54 del Reglamento de la Ley General de Salud en Materia de Control Sanitario de la Disposición de Órganos, Tejidos y Cadáveres de Seres Humanos, así como 3o, fracción I, literal a y 10, fracciones IV y VIII del Reglamento de la Comisión Federal para la Protección contra Riesgos Sanitarios, he tenido a bien ordenar la publicación en el Diario Oficial de la Federación, del
PROYECTO DE NORMA OFICIAL MEXICANA NOM-253-SSA1-2024, PARA LA DISPOSICIÓN DE SANGRE HUMANA Y SUS COMPONENTES CON FINES TERAPÉUTICOS.
El presente proyecto se publica a efecto de que los interesados, dentro de los 60 días naturales siguientes al de la fecha de su publicación en el Diario Oficial de la Federación y Plataforma Tecnológica Integral de Infraestructura de la Calidad, presenten sus comentarios en idioma español ante el Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, ubicado en Oklahoma número 14, planta baja, colonia Nápoles, código postal 03810, Demarcación Territorial Benito Juárez, Ciudad de México, teléfono 50805200, extensión 1333, correo electrónico rfs@cofepris.gob.mx.
Durante el plazo mencionado y de conformidad con lo dispuesto en el artículo 35, fracción V de la Ley de la Infraestructura de la Calidad, los documentos que sirvieron de base para la elaboración del presente proyecto y el Análisis de Impacto Regulatorio, estarán a disposición del público en general, para su consulta, en el domicilio del mencionado Comité, en tanto no se hayan emitidos los lineamientos que regularán el acceso a la Plataforma Tecnológica Integral de Infraestructura de la Calidad por parte de los interesados, y los formatos electrónicos que deberán utilizarse para esos efectos.
CONSIDERANDO
Que la Constitución Política de los Estados Unidos Mexicanos en su artículo 4o, reconoce el derecho humano que toda persona tiene a la protección de la salud; por lo que el Estado tiene la obligación de garantizar y establecer los mecanismos necesarios para que toda persona goce de un estado de completo bienestar físico, mental y social para su desarrollo;
Que el artículo 3o, fracciones XVI y XXVI de la Ley General de Salud establece que son materias de salubridad general, la prevención y el control de enfermedades no transmisibles, sindemias y accidentes y el control sanitario de la disposición de órganos, tejidos y sus componentes y células, entre otros;
Que el artículo 10 de la Ley de Infraestructura de la Calidad, establece que las Normas Oficiales Mexicanas tienen como finalidad atender las causas de los problemas identificados por las Autoridades Normalizadoras que afecten o que pongan en riesgo los objetivos legítimos de interés público, considerados entre otros, como objetivos legítimos de interés público, la protección y promoción a la salud y cualquier otra necesidad pública, en términos de las disposiciones legales aplicables;
Que con fecha 26 de octubre de 2012, se publicó en el Diario Oficial de la Federación la Norma Oficial Mexicana NOM- 253-SSA1-2012, Para la disposición de sangre humana y sus componentes con fines terapéuticos, la cual tiene como objetivo establecer las actividades, criterios, estrategias y técnicas operativas del Sistema Nacional de Salud, en relación con la disposición de sangre humana y sus componentes con fines terapéuticos, y
Que se considera necesario modificar la Norma Oficial Mexicana NOM-253-SSA1-2012, para la disposición de sangre humana y sus componentes con fines terapéuticos, a efecto de alinearse a avances científicos y tecnológicos. Desde la publicación de la norma en el año 2012, ha habido avances significativos en el conocimiento de la sangre, sus componentes y las enfermedades transmisibles por transfusión. Las pruebas de detección de patógenos se han vuelto más sensibles y específicas, y han surgido nuevas tecnologías relacionadas con la sangre. La actualización de la norma permitiría incorporar estos avances para garantizar la seguridad y eficacia de la sangre y sus componentes. se debe además de generar una alineación con estándares internacionales en materia de seguridad transfusional; esto facilitaría la colaboración y el intercambio de información con otros países, así como la adopción de mejores prácticas a nivel global. Por otra parte, se optimizarán procesos mediante la experiencia adquirida en la aplicación de la norma, ya que esto ha revelado áreas de oportunidad para mejorar la eficiencia y eficacia de los procesos relacionados con la disposición de sangre y sus componentes, reduciendo costos y mejorando la calidad de los servicios. También la adaptación a nuevas necesidades derivadas de los cambios demográficos y epidemiológicos han generado nuevas necesidades en materia de sangre y componentes sanguíneos; la actualización de la norma permitiría adaptarse a estas nuevas necesidades, asegurando la disponibilidad de sangre segura y suficiente para todos los pacientes que la requieran y finalmente se generará un fortalecimiento de la seguridad transfusional al reforzar las medidas de seguridad en todas las etapas del proceso, desde la selección de donantes hasta la transfusión de sangre y componentes. Esto contribuiría a reducir aún más el riesgo de transmisión de enfermedades y a garantizar la seguridad de los pacientes, minimizando los efectos adversos a la transfusión.
PREFACIO
En la elaboración del presente Proyecto de Norma participaron las unidades administrativas e instituciones siguientes:
SECRETARÍA DE SALUD
Dirección General de Epidemiología
Centro Nacional de la Transfusión Sanguínea
Centro Nacional para la Prevención y el Control del VIH/SIDA
Comisión Coordinadora de los Institutos Nacionales de Salud y Hospitales de Alta Especialidad
Comisión Federal para la Protección contra Riesgos Sanitarios
Comisión de Control Analítico y Ampliación de Cobertura
Centro Nacional de Excelencia Tecnológica en Salud
Laboratorios de Biológicos y Reactivos de México, S.A. de C.V.
SECRETARÍA DE SALUD DEL GOBIERNO DEL ESTADO DE BAJA CALIFORNIA SUR
Centro Estatal de la Transfusión Sanguínea
SECRETARÍA DE SALUD DEL GOBIERNO DEL ESTADO DE DURANGO
Centro Estatal de la Transfusión Sanguínea
SECRETARÍA DE SALUD DEL GOBIERNO DEL ESTADO DE PUEBLA
Centro Estatal de la Transfusión Sanguínea
SECRETARÍA DE SALUD DEL GOBIERNO DEL ESTADO DE SAN LUIS POTOSÍ
Centro Estatal de la Transfusión Sanguínea
SECRETARÍA DE SALUD DEL GOBIERNO DEL ESTADO DE SONORA
Centro Estatal de la Transfusión Sanguínea
SECRETARÍA DE LA DEFENSA NACIONAL
Dirección General de Sanidad Militar
SECRETARÍA DE MARINA
Dirección General de Sanidad Naval
INSTITUTO MEXICANO DEL SEGURO SOCIAL
Dirección de Prestaciones Médicas
Coordinación de Unidades Médicas de Alta Especialidad
INSTITUTO DE SEGURIDAD Y SERVICIOS SOCIALES DE LOS TRABAJADORES DEL ESTADO
Subdirección General Médica
PETRÓLEOS MEXICANOS
Subdirección Corporativa de Servicios Médicos
INSTITUTO POLITÉCNICO NACIONAL
Escuela Nacional de Ciencias Biológicas
CRUZ ROJA MEXICANA, IAP
ASOCIACIÓN MEXICANA DE MEDICINA TRANSFUSIONAL, A.C.
PLASMA QUE SALVA, A.C.
ÍNDICE
0 Introducción
1 Objetivo y campo de aplicación
2 Referencias normativas
3 Términos y definiciones
4 Símbolos y términos abreviados
5 Disposiciones generales
6 Información, consentimientos y atención para donantes y receptores
7 Selección de donantes para uso terapéutico alogénico
8 Extracción de unidades de sangre y productos sanguíneos para uso alogénico
9 Procesamiento, conservación, vigencia y control de calidad de las unidades de sangre y productos sanguíneos
10 Determinaciones analíticas
11 Identificación de las unidades y de las muestras sanguíneas
12 Selección de unidades de sangre y productos sanguíneos para uso transfusional
13 Disposición de sangre y productos sanguíneos para uso autólogo
14 Solicitudes de transfusión, suministro y recepción, traslado y readmisión de unidades de sangre y productos sanguíneos
15 Transfusión de unidades y reacciones adversas a la transfusión
16 Evaluación de la conformidad y control de calidad
17 Destino final de las unidades de sangre, productos sanguíneos y de las muestras
18 Comité de medicina transfusional
19 Información relativa a la disposición de sangre y productos sanguíneos a la Secretaría de Salud
20 Procedimientos normalizados de operación, guías, instructivos, documentos y registros
21 Concordancia con normas internacionales y mexicanas
22 Bibliografía
23 Observancia de la norma
24 Vigilancia de la norma
25 Vigencia
APÉNDICE A NORMATIVO Procedimientos terapéuticos para diversos padecimientos
APÉNDICE B NORMATIVO Preceptos de control de calidad
APÉNDICE C NORMATIVO Embalaje y transporte para conservación de sangre y productos sanguíneos
0 Introducción
La Organización Mundial de la Salud y la Organización Panamericana de la Salud establecen que para abastecer de sangre segura a la población se debe fomentar el trabajo en equipo, obtener la sangre y productos sanguíneos de donantes voluntarios y altruistas, no remunerados y regulares, asegurándose que reciban una atención de calidad.
A la par deben establecerse programas para una evaluación estricta de los donantes, así como para el procesamiento fraccionamiento, conservación, análisis, suministro y aplicación terapéutica de los productos sanguíneos.
Todos los productos sanguíneos colectados deben ser estudiados para la detección de marcadores de agentes infecciosos transmisibles por transfusión, tales como el virus de la inmunodeficiencia humana, los virus B y C de la hepatitis, Trypanosoma cruzi, Treponema pallidum y otros que según diversas circunstancias se hagan necesarios.
Con el fin de garantizar la autosuficiencia, cobertura universal y seguridad de la sangre y sus componentes, debe actualizarse el marco jurídico en la materia, fomentar una coordinación eficiente de los servicios de sangre del país, con criterios de integración en un sistema nacional de sangre con redes de atención, así como, promover la donación voluntaria, no remunerada y regular como una fuente segura de obtención de la sangre y productos sanguíneos; implementar técnicas de laboratorio con mayor sensibilidad y especificidad y fomentar el uso adecuado y racional de la sangre total y de los productos sanguíneos.
Esta Norma debe contribuir a la confianza general en cuanto a la donación de sangre y productos sanguíneos, dando protección a la salud de los donantes, receptores y el personal de salud, conseguir la autosuficiencia, reforzar la seguridad de la cadena transfusional, de manera suficiente y que pueda lograrse un mejor nivel de atención, adoptando las medidas necesarias para alcanzar los objetivos planteados.
La donación voluntaria no remunerada y regular, la selección adecuada del donante y el mejoramiento de las pruebas de laboratorio, han permitido que en las últimas dos décadas hubiera una reducción importante del riesgo de transmisión transfusional de agentes infecciosos. Con el fin de disminuir los riesgos de transmisión de agentes infecciosos transmisibles por transfusión, este proyecto de Norma actualiza las metodologías de laboratorio con pruebas más sensibles y específicas que se aplican a los donantes.
Con el fin de incrementar la seguridad transfusional, se instauran las bases para la biovigilancia y la hemovigilancia, programas que proporciona información útil acerca de la morbilidad y mortalidad en torno a la donación sanguínea y a la transfusión, al tiempo que constituye una guía sobre las medidas preventivas para evitar o disminuir eventos y reacciones adversas a la donación y a la transfusión. La biovigilancia y la hemovigilancia posibilitan que de manera inmediata se activen los mecanismos de alerta y correctores necesarios ante cualquier complicación atribuible a la donación o a la transfusión. Esta información garantiza que se establezca un control de calidad continuo de la cadena transfusional, hecho que reporta beneficios indiscutibles, tanto para los donantes como para los receptores de sangre y productos sanguíneos.
De igual manera se consideran las determinantes sociales de la cadena transfusional con una perspectiva universal, equitativa, empática y no discriminatoria. Es decir, incluyente de género, religión, pueblos originarios, migrantes, personas con discapacidad y/o trastornos mentales, bajo los principios de participación social, competencia técnica y calidad de la atención médica con la intención de "No dejar a nadie atrás, no dejar a nadie afuera".
1 Objetivo y campo de aplicación
1.1 Este proyecto de Norma tendrá por objeto establecer las actividades, criterios, estrategias y técnicas operativas del Sistema Nacional de Salud, en relación con la disposición de sangre humana y sus componentes con fines terapéuticos.
La regulación de los hemoderivados, tales como la albúmina, las inmunoglobulinas, los concentrados de factores de coagulación, entre otros, obtenidos mediante procedimientos fisicoquímicos o biológicos, serán materia de otras disposiciones.
1.2 Este proyecto de Norma será de observancia obligatoria en todo el territorio nacional para todo el personal profesional, técnico, auxiliar y administrativo de los establecimientos públicos, sociales y privados que hacen disposición de sangre humana y sus componentes con fines terapéuticos.
2 Referencias normativas
Para la aplicación correcta de este Proyecto de Norma es necesario consultar las siguientes normas oficiales mexicanas o las que la sustituyan.
2.1 NORMA Oficial Mexicana NOM-002-STPS-2010, Condiciones de seguridad-prevención y protección contra incendios en los centros de trabajo.
2.2 NORMA Oficial Mexicana NOM-004-SSA3-2012, Del expediente clínico.
2.3 NORMA Oficial Mexicana NOM-005-SSA3-2018, Que establece los requisitos mínimos de infraestructura y equipamiento de establecimientos para la atención medica de pacientes ambulatorios.
2.4 NORMA Oficial Mexicana NOM-010-SSA-2023, Para la prevención y el control de la infección por virus de la inmunodeficiencia humana.
2.5 NORMA Oficial Mexicana NOM-012-STPS-2012, Condiciones de seguridad y salud en los centros de trabajo donde se manejen fuentes de radiación ionizante.
2.6 NORMA Oficial Mexicana NOM-015-SSA2-2010, Para la prevención, tratamiento y control de la diabetes mellitus.
2.7 NORMA Oficial Mexicana NOM-016-SSA3-2012, Que establece las características mínimas de infraestructura y equipamiento de hospitales y consultorios de atención médica especializada.
2.8 NORMA Oficial Mexicana NOM-017-STPS-2008, Equipo de protección personal-selección, uso y manejo en los centros de trabajo
2.9 NORMA Oficial Mexicana NOM-017-SSA2-2012, Para la vigilancia epidemiológica.
2.10 NORMA Oficial Mexicana NOM-024-SSA3-2012, Sistemas de información de registro electrónico para la salud. Intercambio de información en salud.
2.11 NORMA Oficial Mexicana NOM-032-SSA2-2014, Para la vigilancia epidemiológica, prevención y control de enfermedades transmitidas por vectores.
2.12 NORMA Oficial Mexicana NOM-035-SSA3-2012, En materia de información en salud.
2.13 Norma Oficial Mexicana NOM-039-SSA2-2014, Para la prevención y control de las infecciones de transmisión sexual.
2.14 NORMA Oficial Mexicana NOM-045-SSA2-2005, Para la vigilancia epidemiológica, prevención y control de las infecciones nosocomiales.
2.15 NORMA Oficial Mexicana NOM-051-SCFI/SSA1-2010, Especificaciones generales de etiquetado para alimentos y bebidas no alcohólicas preenvasados-Información comercial y sanitaria.
2.16 NORMA Oficial Mexicana NOM-052-SEMARNAT-2005, Que establece las características, el procedimiento de identificación, clasificación y los listados de los residuos peligrosos.
2.17 NORMA Oficial Mexicana NOM-059-SSA1-2015, Buenas prácticas de fabricación de medicamentos.
2.18 NORMA Oficial Mexicana NOM-064-SSA1-1993, Que establece las especificaciones sanitarias del equipo de reactivos utilizados para diagnóstico.
2.19 NORMA Oficial Mexicana NOM-078-SSA1-1994, Que establece las especificaciones sanitarias de los estándares de calibración utilizados en las mediciones realizadas en los laboratorios de patología clínica.
2.20 NORMA Oficial Mexicana NOM-087-SEMARNAT/SSA1-2002, Protección ambiental - Salud ambiental - Residuos peligrosos - biológico - infecciosos - Clasificación y especificaciones de manejo.
2.21 NORMA Mexicana NMX-CC-9000-IMNC-2015. Sistemas de gestión de calidad-fundamentos y vocabulario.
2.22 NORMA Mexicana NMX-CC-9001-IMNC-2008, Sistemas de gestión de la calidad-requisitos.
2.23 NORMA Mexicana NMX-EC-15189-IMNC-2015. Requisitos particulares para la calidad y la competencia de los laboratorios clínicos.
2.24 NORMA Mexicana NMX-EC-17043-IMNC-2010, Evaluación de la conformidad-requisitos generales para los ensayos de aptitud.
2.25 NORMA Mexicana NMX-Z-005-IMNC-2009, Vocabulario Internacional de metrología-conceptos fundamentales y generales asociados (VIM).
3 Términos y definiciones
3.1 Para los fines de esta Norma son aplicables las definiciones siguientes:
3.1.1 Acción correctiva: a las actividades que son planeadas y ejecutadas, con el fin de eliminar la causa de una desviación o no conformidad.
3.1.2 Acción preventiva: a las actividades que son planeadas y ejecutadas, para eliminar la causa de una desviación o no conformidad u otra situación potencialmente indeseable y evitar su recurrencia.
3.1.3 Acreditación: el acto por el cual una entidad de acreditación reconoce la competencia técnica y confiabilidad de los organismos de certificación, de los laboratorios de prueba, de los laboratorios de calibración y de las unidades de verificación para la evaluación de la conformidad.
3.1.4 Ácido desoxirribonucleico (ADN): molécula que contiene la información genética de un individuo que se hereda de generación en generación y funciona como almacenamiento de información para el mantenimiento, división y construcción de otros componentes celulares.
3.1.5 Aféresis: procedimiento que tiene por objeto la separación de componentes de la sangre provenientes de un solo donante, mediante centrifugación, por medio de circuitos cerrados y estériles en equipos de flujo continuo o discontinuo.
3.1.6 Agente biológico-infeccioso: cualquier microorganismo capaz de producir enfermedades cuando está presente en concentraciones suficientes (inóculo), en un ambiente propicio (supervivencia), en un hospedero susceptible y en presencia de una vía de entrada.
3.1.7 Aglutinación: reacción caracterizada por agrupación de células o partículas resultante de la interacción entre antígenos y anticuerpos.
3.1.8 Alelo: es una de dos o más versiones de una secuencia del ácido desoxirribonucleico (una base única o un segmento de bases) en una ubicación genómica determinada.
3.1.9 Aloanticuerpo: inmunoglobulina resultante de una respuesta inmune a un antígeno ajeno al individuo.
3.1.10 Aloinmunización: producción de anticuerpos por un individuo dirigidos contra de otro individuo de la misma especie.
3.1.11 Anemia: concentración de hemoglobina en sangre inferior al valor esperado, teniendo en cuenta la edad, el sexo.
3.1.12 Anticuerpo: inmunoglobulina resultante de una respuesta inmune a un antígeno propio o ajeno al individuo.
3.1.13 Anticuerpo irregular de importancia clínica: inmunoglobulina plasmática poco frecuente (prevalencia menor del 1%) que puede causar enfermedad a través de diferentes mecanismos.
3.1.14 Antígeno: sustancia capaz de estimular una respuesta inmune con la formación de anticuerpos.
3.1.15 Autonomía frigorífica: el tiempo que transcurre desde que se cargan o introducen en un contenedor los bloques refrigerantes congelados, hasta que la temperatura interior máxima alcanza +10 º C, con una temperatura exterior constante de +43 º C, y siempre que el contenedor se mantenga cerrado.
3.1.16 Bacteremia: presencia de bacterias en el torrente sanguíneo.
3.1.17 Bancos de Sangre: establecimiento autorizado para obtener, recolectar, analizar, procesar, conservar, aplicar y proveer sangre humana; así como para analizar, conservar, aplicar y proveer los componentes de la misma.
3.1.18 Biovigilancia: Proceso para garantizar la calidad y seguridad de las células y los tejidos a través del registro y la transmisión de información sobre los efectos y reacciones adversas debido a alguno de los procesos desde la donación hasta el trasplante.
3.1.19 Buenas prácticas de fabricación: al conjunto de lineamientos y actividades relacionadas entre sí, destinadas a asegurar que los medicamentos elaborados tengan y mantengan las características de identidad, pureza, seguridad, eficacia y calidad requeridas para su uso.
3.1.20 Calibración: a la demostración de que un instrumento particular o dispositivo produce resultados dentro de límites especificados, en comparación con los producidos por una referencia o estándar trazable sobre un intervalo de mediciones establecido.
3.1.21 Calidad: al cumplimiento de especificaciones establecidas para garantizar la aptitud de uso.
3.1.22 Candidata (o) a donar: persona cuya aptitud para donar sangre o productos sanguíneos será evaluada por un médico capacitado para dicho fin.
3.1.23 Capa leucocitaria: fracción sanguínea que contiene principalmente leucocitos, separada por centrifugación de una unidad de sangre total.
3.1.24 Capa leucoplaquetaria: fracción sanguínea que contiene principalmente leucocitos y plaquetas, separada por centrifugación de una unidad de sangre total.
3.1.25 Centigray (cGy): la centésima parte de un gray (Gy).
3.1.26 Centro de Procesamiento de Sangre: establecimiento autorizado para recibir sangre y sus componentes provenientes de los centros de colecta para la preparación, análisis y conservación de los mismos, así como el envío de productos sanguíneos y muestras a los servicios de transfusión hospitalario y centros de distribución de sangre y productos sanguíneos. Se encuentra bajo la responsabilidad de un banco de sangre.
3.1.27 Centro de Colecta: establecimiento autorizado para hacer la recolección y obtención de sangre total y sus componentes mediante procedimiento de aféresis, contemplando la atención al donador, resguardo y envío de sangre y/o productos sanguíneos obtenidos para su análisis en un centro de procesamiento de sangre o banco de sangre. Se encuentra bajo la responsabilidad de un banco de sangre.
3.1.28 Centro de Distribución de Sangre y Productos sanguíneos: establecimiento autorizado para la recepción de productos sanguíneos provenientes de bancos de sangre y de centros de procesamiento de sangre, además de su conservación y envío a servicios de transfusión hospitalario y bancos de sangre. Se encuentra bajo la responsabilidad de un banco de sangre.
3.1.29 Centro de Calificación Biológica: establecimiento público autorizado para realizar pruebas de inmunohematología, amplificación de ácidos nucleicos y determinaciones analíticas para la detección de agentes infecciosos transmisibles por transfusión, así como la realización de las pruebas confirmatorias y suplementarias. Se encuentra bajo la responsabilidad de un banco de sangre público.
3.1.30 Certificación: procedimiento por el cual se asegura que un producto, proceso, sistema o servicio, se ajusta a las normas, lineamientos o recomendaciones de organismos dedicados a la normalización nacionales o internacionales.
3.1.31 Citaféresis: procedimiento mecánico por el cual se extrae selectivamente de un donante una o más líneas celulares de la sangre y transfunde el remanente al propio donante.
3.1.32 Clona: copia idéntica de un organismo, célula o molécula.
3.1.33 Colecta externa: proceso de recolección de sangre y productos sanguíneos, realizado fuera de las instalaciones del servicio de sangre que cuenta con licencia sanitaria vigente.
3.1.34 Comité de Medicina Transfusional: grupo constituido por un número variable de profesionales de la salud de acuerdo con los servicios de atención médica, tamaño y grado de especialización del hospital, cuya responsabilidad es asegurar la calidad y seguridad del ejercicio transfusional.
3.1.35 Complejos de anticuerpos múltiples: una muestra que contiene cuatro o más aloanticuerpos contra antígenos celulares.
3.1.36 Componente sanguíneo: fracción celular o acelular del tejido hemático, separada de una unidad de sangre total por centrifugación u obtenida por aféresis, también llamado hemocomponentes.
3.1.37 Control de calidad: son las actividades y técnicas operativas desarrolladas para cumplir con los requisitos de calidad establecidos.
3.1.38 Control de calidad externo: es la evaluación realizada periódicamente por un proveedor de ensayos de aptitud reconocido por una entidad de acreditación, de los análisis o ensayos que efectúa un establecimiento y que tiene por objeto verificar que las técnicas, reactivos, procedimientos e interpretación de los resultados son los correctos.
3.1.39 Control de calidad interno: el proceso que tiene por objeto, a través de pruebas realizadas cada vez que se efectúa un análisis o ensayo o conjunto de ensayos de la misma técnica, para detectar y corregir errores eventuales.
3.1.40 Corrida: procedimiento de laboratorio en el que en una sesión se incluyen para su análisis diferentes muestras sanguíneas, habitualmente suero o plasma, empleando el mismo método, reactivos, controles, equipos e instrumentos.
3.1.41 Crioprotección: métodos empleados para la salvaguarda de la viabilidad de cualquier tipo de células al someterlas a congelación (-70º C) o ultracongelación (-150º C).
3.1.42 Cuarentena: aislamiento físico de los productos sanguíneos, materiales y reactivos durante un periodo de tiempo variable, en espera de su aceptación, suministro o rechazo.
3.1.43 Daño pulmonar agudo asociado a transfusión (TRALI, por sus siglas en inglés): síndrome de presentación súbita caracterizado por disnea, hipoxemia e infiltrados pulmonares intersticiales, que se presenta durante o en el lapso de las primeras seis horas tras una transfusión, en ausencia de otras causas detectables.
3.1.44 Depósito previo: acto de disposición para uso autólogo en el que anticipadamente a su empleo terapéutico, se acopia la sangre o sus componentes.
3.1.45 Disposición de sangre: conjunto de actividades relativas a la obtención, recolección, análisis, conservación, preparación, suministro, utilización y destino final de la sangre y productos sanguíneos, con fines terapéuticos.
3.1.46 Donador (o donante): al que tácita o expresamente consiente la disposición en vida de su sangre, productos sanguíneos y células progenitoras.
3.1.47 Donador autólogo: la persona que proporciona sangre para uso exclusivo en sí misma, cuando se anticipa el requerimiento transfusional y se desarrolla un plan de donación.
3.1.48 Donador designado: la persona en la que existe una clara indicación médica para el uso de su sangre o productos sanguíneos en un paciente determinado.
3.1.49 Donador dirigido: la persona que por su voluntad pretende que su sangre o componentes de ésta sean utilizados en algún paciente determinado.
3.1.50 Donador familiar o de reposición: persona que proporciona su sangre o productos sanguíneos a favor de un paciente, en respuesta a una solicitud específica por parte del personal de salud, familiares o amigos del paciente.
3.1.51 Donador regular: la persona que ha proporcionado sangre o cualquier componente sanguíneo en más de una ocasión en el lapso de los últimos dos años en el mismo centro de colecta.
3.1.52 Donador de repetición: la persona que proporciona su sangre o productos sanguíneos en dos o más ocasiones en el lapso de un año.
3.1.53 Donador voluntario y altruista: persona que proporciona su sangre o productos sanguíneos para uso terapéutico de quien lo requiera, sin la intención de beneficiar a una persona en particular, motivada únicamente por sentimientos humanitarios y de solidaridad, sin esperar retribución alguna a cambio y sin que medie una solicitud específica por parte del personal de salud, familiares o amigos del paciente.
3.1.54 Eluido: medio fluido que contiene anticuerpos que fueron separados deliberadamente de un antígeno celular.
3.1.55 Especificidad: probabilidad de una prueba de laboratorio para identificar todos los negativos o no reactivos correctamente.
3.1.56 Esterilización: eliminación, inactivación o la destrucción de todas las formas de vida microbiana por medio del uso de agentes químicos o físicos.
3.1.57 Evaluación de la conformidad: la determinación del grado de cumplimiento con las normas oficiales mexicanas o la conformidad con las normas mexicanas, las normas internacionales u otras especificaciones, prescripciones o características. Comprende, entre otros, los procedimientos de muestreo, prueba, calibración, certificación y verificación.
3.1.58 Evento de riesgo: suceso imprevisto o de realización insegura que podría llevar a un resultado adverso.
3.1.59 Exsanguineotransfusión: procedimiento terapéutico que consiste en cambiar la sangre de una persona, sustituyéndola por sangre reconstituida proveniente de donadores cuyos eritrocitos y plasma conserven todas sus propiedades terapéuticas.
3.1.60 Factor de riesgo: condición que incrementa la probabilidad de desarrollar enfermedad o alteración de la salud.
3.1.61 Fecha de caducidad o límite de vigencia: el último día en que las unidades de sangre, productos sanguíneos, los materiales, las sustancias y los reactivos se consideran viables o útiles.
3.1.62 Fenotipo: son todos los rasgos observables de un individuo como el color de ojos, sistema de grupo sanguíneo, estatura, etcétera. Se determina por la composición genómica y factores ambientales.
3.1.63 Gestión de la calidad: conjunto de actividades coordinadas para dirigir y controlar una organización en lo que respecta a la calidad en todos los niveles del establecimiento.
3.1.64 Gray (Gy): unidad de dosis absorbida de energía ionizante, equivalente a 100 rads.
3.1.65 Genotipo: es toda la composición genómica (codificante o no codificante) de un individuo que se hereda de generación en generación según los patrones de herencia.
3.1.66 Genotipificación sanguínea: prueba realizada a partir del ácido desoxirribonucleico que identifica las variantes genéticas de las glicoproteínas de superficie membranal del eritrocito o de las plaquetas, permitiendo la predicción de un determinado sistema de grupo sanguíneo o de antígenos plaquetarios humanos.
3.1.67 Hemoclasificación: sistema de clasificación de antígenos (ausentes o presentes) de la superficie de membrana del eritrocito que determina el sistema de grupo sanguíneo de cada persona.
3.1.68 Hemoderivados: los productos obtenidos de algunos productos sanguíneos, especialmente el plasma, mediante procesos fisicoquímicos o biológicos, para aplicación terapéutica, diagnóstica, preventiva o en investigación.
3.1.69 Hemodilución aguda preoperatoria: acto de disposición para uso autólogo en el que se colecta sangre en el preoperatorio inmediato, manteniendo el volumen sanguíneo circulatorio con la administración de soluciones.
3.1.70 Hemovigilancia: conjunto de procedimientos organizados para dar seguimiento a los efectos o reacciones adversas o inesperadas que se manifiestan en los donadores o en los receptores, con el fin de prevenir su aparición o recurrencia.
3.1.71 Hiperpotasemia (hiperkalemia): exceso de la concentración de potasio en sangre según el valor superior de referencia.
3.1.72 Inactivación en productos sanguíneos: técnicas validadas y estandarizadas consistentes en someter a un componente sanguíneo a un tratamiento in-vitro, con el objeto impedir la transmisión de agentes infecciosos, la enfermedad injerto contra huésped (u hospedero) y otras patologías.
3.1.73 Identificación de anticuerpos: proceso diseñado para conocer la especificidad de uno o varios anticuerpos.
3.1.74 Incidente: evento inesperado, no planeado ni atribuible al error de una persona.
3.1.75 Incompatibilidad mayor: cuando el plasma del receptor contiene anticuerpos en contra de los eritrocitos del donador.
3.1.76 Incompatibilidad menor: cuando el plasma del donador contiene anticuerpos en contra de los eritrocitos del receptor.
3.1.77 Inmunoglobulina: proteína presente en el plasma, de mayor masa molecular que la albúmina, que actúa como anticuerpo.
3.1.78 Inmunohematología: el estudio de los antígenos y anticuerpos de los grupos sanguíneos y problemas asociados.
3.1.79 In vitro: estudios o experimentos que se realizan fuera de un organismo biológico.
3.1.80 Irradiación: procedimiento en el que se somete un componente celular de la sangre a la acción de radiación ionizante por métodos previamente estandarizados y autorizados, con la finalidad de evitar en el receptor la enfermedad injerto contra huésped (u hospedero) asociada a transfusión.
3.1.81 Isogrupo: se refiere al sistema de grupo sanguíneo, por ejemplo, ABO.
3.1.82 Isohemaglutininas: son proteínas en contra de antígenos de los sistemas de grupo sanguíneo que generan hemaglutinación.
3.1.83 Liberación: al estado de la sangre y productos sanguíneos, que permiten su disposición para su uso terapéutico, posterior al estado en que puede evidenciarse que cumplen con los requisitos de calidad establecidos.
3.1.84 Linfotoxicidad: proceso celular relacionado a los efectos tóxicos de los linfocitos.
3.1.85 Leucodepleción: procedimiento por el cual se disminuyen de tres o más logaritmos los leucocitos de algún componente celular de la sangre; se logra con el empleo de filtros de última generación.
3.1.86 Leucorreducción: procedimiento por el cual se disminuyen hasta un logaritmo los leucocitos de la sangre o algún componente celular de la sangre; puede lograrse con los métodos actuales de procesamiento.
3.1.87 Marbete: información contenida en una etiqueta que acompaña a una unidad de sangre o de algún componente sanguíneo, pero que no va adherida a la unidad.
3.1.88 Mejora continua: actividad recurrente para aumentar la capacidad de cumplir los requisitos mediante el establecimiento de objetivos y a través de los hallazgos de la auditoría, el análisis de datos, la revisión por la dirección u otros medios que conducen a la acción correctiva.
3.1.89 Mezcla de componentes: volumen resultante de combinar, en condiciones de esterilidad, dos o más unidades de productos sanguíneos.
3.1.90 Muestra: alícuota de sangre, plasma, suero o de un producto extraída del conjunto por métodos que permitan considerarla como representativa del mismo, empleada para fines de diagnóstico, comprobación o investigación, no utilizable para fines terapéuticos.
3.1.91 PAAN: se refiere a pruebas de amplificación de ácidos nucleicos. Cuando se indica PAAN, se refiere a la implementación de pruebas moleculares de escrutinio para la detección de virus de inmunodeficiencia humana y los virus para Hepatitis B y C.
3.1.92 Paciente: a todo aquel usuario beneficiario directo de la atención médica.
3.1.93 Paciente con poliglobulia: persona que por un proceso patológico primario o secundario, tiene un incremento absoluto del volumen eritrocítico circulante.
3.1.94 Paraproteinemia: presencia en plasma o suero de proteínas anormales o en cantidad excesiva.
3.1.95 Periodo de ventana: el lapso entre el momento del contagio con un agente infeccioso y el desarrollo de marcadores de infección detectables en el suero de una persona.
3.1.96 Pirógeno: es una sustancia que provoca un aumento de la temperatura en un ser humano o un animal a través de la activación del sistema inmunitario.
3.1.97 Prion: agente infeccioso constituido por un péptido mal plegado, que produce alteraciones neurodegenerativas contagiosas en el ser humano y en diversas especies animales.
3.1.98 Procedimiento normalizado de operación: documento que contiene las instrucciones necesarias para llevar a cabo de manera reproducible una actividad.
3.1.99 Proceso crítico: actividad o conjunto de actividades cuya metodología de ejecución pueda afectar significativamente la seguridad de los donadores, los receptores, los productos sanguíneos o los servicios prestados, que pueda influir en la calidad del producto final y en los servicios prestados.
3.1.100 Productos sanguíneos: término genérico empleado para designar los diversos preparados de la sangre que tienen utilidad terapéutica, incluyen las unidades de sangre total, de sus componentes y mezclas de éstos.
Los diversos productos sanguíneos se definen, en orden lógico, en la tabla 1 de este proyecto de Norma.
Tabla 1
Definición de los productos sanguíneos
Sangre |
3.1.101 Sangre: el tejido hemático con todos sus componentes. 3.1.102 Sangre total: el tejido hemático tal y como se obtiene en una sesión de extracción, suspendido en una solución anticoagulante. 3.1.103 Sangre fresca: el tejido hemático de reciente extracción, que se ha mantenido en condiciones adecuadas de conservación y que mantiene todas las propiedades de sus diversos componentes, empleada para la obtención de diversos productos sanguíneos. 3.1.104 Sangre reconstituida: unidad de concentrado de eritrocitos a la que se le agrega plasma en cantidad suficiente para obtener un hematocrito dentro del rango normal. 3.1.105 Sangre reconstituida unitaria: el concentrado de eritrocitos al que se le ha agregado su propio plasma fresco descongelado hasta lograr un hematocrito útil para fines terapéuticos. 3.1.106 Sangre reconstituida de distintos donadores: el concentrado de eritrocitos al que se le ha agregado plasma fresco descongelado, proveniente de otro donador, hasta lograr un hematocrito útil para fines terapéuticos. |
Productos sanguíneos |
3.1.107 Componente sanguíneo: fracción celular o acelular del tejido hemático, separada de una unidad de sangre total por centrifugación u obtenida por aféresis, también llamado hemocomponentes. 3.1.108 Componente acelular: unidad o mezcla de productos sanguíneos carente de elementos celulares, que contiene plasma o algún componente plasmático. 3.1.109 Componente celular: unidad o mezcla de productos sanguíneos que contiene alguna fracción o fracciones de la sangre con alto contenido de uno o más elementos celulares. |
Concentrados de eritrocitos |
3.1.110 Concentrado de eritrocitos: unidad que contiene mayoritariamente glóbulos rojos, obtenidos por procesamiento de una unidad de sangre total de una donación única o de una sesión de eritroaféresis. 3.1.111 Concentrado de eritrocitos en solución aditiva: unidad que contiene mayoritariamente glóbulos rojos, obtenidos por procesamiento de una unidad de sangre total proveniente de una donación única o de una sesión de aféresis a la que se añade una solución nutritiva o conservadora. 3.1.112 Concentrado de eritrocitos en solución aditiva sin la capa leucoplaquetaria: unidad de glóbulos rojos de la que se ha eliminado gran parte la capa donde se localizan los leucocitos y las plaquetas. 3.1.113 Concentrado de eritrocitos leucodepletado: unidad de glóbulos rojos sometida a eliminación de leucocitos hasta una cifra igual o menor de un millón por unidad, mediante técnicas de filtración. 3.1.114 Concentrados de eritrocitos lavados: unidad de glóbulos rojos de la que se han removido en proporción suficiente el plasma y la capa leucoplaquetaria mediante enjuagues sucesivos con solución salina isotónica. 3.1.115 Concentrado de eritrocitos congelados: unidad de glóbulos rojos en una solución de glicerol, como agente crioprotector, que permite conservarlos a temperaturas de congelación de -70º C o menores, e incrementar su periodo de vigencia. 3.1.116 Concentrado de eritrocitos irradiados: unidad de glóbulos rojos sometida a técnicas estandarizadas de radiación ionizante. |
Preparados con plaquetas |
3.1.117 Concentrado de plaquetas: unidad que contiene principalmente trombocitos suspendidos en plasma o solución aditiva que son obtenidos por aféresis o preparados mediante procesamiento de unidades de sangre fresca de una donación única. 3.1.118 Concentrado de plaquetas unitario o recuperado: unidad que contiene trombocitos en suspensión, obtenida mediante procesamiento de una unidad de sangre total. 3.1.119 Mezcla de plaquetas: volumen por dosis, resultante de combinar en condiciones de esterilidad varias unidades de concentrados de plaquetas. 3.1.120 Concentrado de plaquetas obtenidas por aféresis: unidad que contiene trombocitos en suspensión obtenida por métodos de aféresis. 3.1.121 Concentrado de plaquetas leucodepletado: unidad o mezcla de trombocitos sometidas a eliminación de glóbulos blancos hasta una cifra igual o menor de un millón por unidad, mediante técnicas de filtración. 3.1.122 Concentrado de plaquetas lavadas: unidad o mezcla con trombocitos recuperados u obtenidos por aféresis, de la que se ha removido en proporción suficiente el plasma mediante enjuagues sucesivos con solución salina isotónica con o sin amortiguador. 3.1.123 Plaquetas irradiadas: unidad o mezcla de plaquetas sometida a técnicas estandarizadas de radiación ionizante. |
Concentrado de granulocitos |
3.1.124 Concentrado de granulocitos: unidad obtenida en una sesión de aféresis, que contiene principalmente neutrófilos suspendidos en plasma. |
Plasmas |
3.1.125 Plasma: el componente específico separado de las células de la sangre. 3.1.126 Plasma fresco: aquel obtenido de un donador de sangre total o mediante aféresis, en estado líquido, mantenido durante un periodo de tiempo y a una temperatura determinada que permitan que los factores lábiles de la coagulación permanezcan funcionales. 3.1.127 Plasma fresco congelado: aquel obtenido de un donador de sangre total o mediante aféresis y que se congela en un periodo de tiempo y a determinada temperatura, que permitan que los factores lábiles de la coagulación se mantengan en estado funcional. 3.1.128 Plasma desprovisto de factores lábiles: aquel que por longevidad o defectos en la conservación ha perdido la actividad de los factores V y VIII de la coagulación. 3.1.129 Plasma desprovisto del crioprecipitado: componente obtenido de una unidad de plasma fresco congelado, consistente en el remanente plasmático que queda al retirar la porción del plasma que precipita en frío. 3.1.130 Plasma rico en plaquetas: aquel que contiene abundantes plaquetas en suspensión, considerando un mínimo de 2,5-1000 x10³ plaquetas/µL suspendidas en plasma. 3.1.131 Plasma en cuarentena: aquel en que se efectúa el control de las pruebas de detección de agentes infecciosos con una nueva determinación en el donador, en tiempo tal que cubra el periodo de ventana habitual de los marcadores de las infecciones virales transmisibles por transfusión. |
Crioprecipitados |
3.1.132 Crioprecipitado: componente que contiene la fracción de crioglobulina precipitada del plasma obtenido mediante el procesamiento posterior del plasma fresco congelado. 3.1.133 Unidad de crioprecipitado: fracción proteica del plasma fresco congelado que precipita al descongelarse en condiciones controladas, obtenida de un solo donador. 3.1.134 Mezcla de crioprecipitados: volumen por dosis resultante de combinar en condiciones de esterilidad varias unidades de crioprecipitados. |
3.1.135 Prueba de antiglobulina humana (PAH) (prueba de Coombs): ensayo de hemaglutinación en el que se emplean anticuerpos contra la gamaglobulina humana, que permite demostrar la presencia o ausencia de anticuerpos adheridos a un antígeno de la membrana del eritrocito.
3.1.136 Prueba de compatibilidad: estudio practicado in vitro empleando muestras de sangre del donador y del receptor, para comprobar la existencia de afinidad inmunológica recíproca entre las células del uno y el suero del otro, para efectos transfusionales.
3.1.137 Prueba de antiglobulina humana directa (PAD) (o Coombs directo): análisis que permite detectar anticuerpos, complemento o ambos, adheridos a la membrana del eritrocito, mediante el uso de anticuerpos contra la gammaglobulina humana y/o contra una fracción de complemento.
3.1.138 Prueba de antiglobulina humana indirecto (PAI) (o Coombs indirecto): análisis que permite detectar en suero o en plasma anticuerpos específicos contra algún antígeno presente en la membrana del eritrocito que se emplee, mediante el uso anticuerpos contra la gammaglobulina humana.
3.1.139 Prueba de tamizaje: análisis presuntivo para la detección de anticuerpos o antígenos de agentes infecciosos transmisibles.
3.1.140 Prueba suplementaria: análisis de laboratorio adicional que apoya los resultados de las pruebas de tamizaje, mas no los confirma.
3.1.141 Radionúclido: el núcleo inestable de un átomo debido a que su proporción de neutrones es mayor o menor al número de protones, y que al tender hacia el equilibrio emite radiación en forma de ondas o partículas.
3.1.142 Reacción o evento adverso: respuesta nociva e inesperada, de aparición inmediata o tardía o incidente, ocurrido en el donador o en el receptor, relacionada con la extracción o la transfusión de sangre o de sus componentes, que ocasiona síntomas, anormalidades, o condiciones temporales o permanentes de diverso grado de severidad.
3.1.143 Reacción o evento adverso grave: respuesta nociva e inesperada o incidente ocurrido en el donador o en el receptor, relacionada con la extracción o la transfusión de sangre o de sus componentes y que resulte mortal, potencialmente mortal, que produzca invalidez o incapacidad o que dé lugar a hospitalización o enfermedad o, en su caso, las prolongue.
3.1.144 Reactivo de antiglobulina humana: antisuero empleado para la detección de globulinas humanas adheridas a los eritrocitos monoespecíficos que contiene solo anti-IgG o poliespecífico que contiene anti-IgG y anti-complemento.
3.1.145 Reactivos hemoclasificadores: antisueros registrados y autorizados que se utilizan para la fenotipificación de la sangre por medio de la identificación de antígenos en la membrana de los eritrocitos.
3.1.146 Recuperación sanguínea: acto de disposición para uso autólogo en el que se colecta la sangre extravasada en el transoperatorio, postoperatorio o ambos.
3.1.147 Refractariedad plaquetaria: respuesta inadecuada a la transfusión de plaquetas después de dos transfusiones consecutivas.
3.1.148 Riesgo: posibilidad o probabilidad de que ocurra una enfermedad o un evento dañino.
3.1.149 Sensibilidad: probabilidad de una prueba de laboratorio para detectar verdaderos reactivos o verdaderos positivos.
3.1.150 Seroteca: espacio donde se almacenan bajo estrictas condiciones de bioseguridad y a temperatura adecuada muestras de suero o plasma, generalmente en alícuotas congeladas, provenientes de donadores, receptores o pacientes, con el fin de efectuar futuras determinaciones analíticas que pudiesen requerirse.
3.1.151 Sistema abierto: se trata de la unidad de sangre total o de algún componente sanguíneo, cuyo interior ha perdido esterilidad, por haberse puesto en contacto con el exterior.
3.1.152 Sistema cerrado: se trata de la unidad de sangre total o algún componente sanguíneo, cuyo interior se mantiene estéril por no haberse puesto en contacto con el exterior o, en su caso, que durante su procesamiento se hubiesen empleado sistemas de conexión estéril.
3.1.153 Sistema de Gestión de la Calidad: un sistema de gestión de la calidad es una estructura operativa de trabajo documentada. Integra los procesos técnicos y gerenciales para guiar las acciones de la fuerza de trabajo, el equipamiento y la información de la organización de manera práctica y coordinada y asegura el cumplimiento de las metas y la satisfacción del usuario. Sus diferentes componentes pueden ser identificados como subsistemas, ya que desde la estructura de la organización se ubican en un orden jerárquico inferior y en conjunto definen y estructuran al sistema.
3.1.154 Servicios de Sangre: establecimientos para la disposición de sangre y productos sanguíneos con fines terapéuticos
3.1.155 Servicios de Transfusión Hospitalarios: establecimiento autorizado para el manejo, conservación, estudios inmunohematológicos y aplicación de sangre humana y sus componentes, obtenidos de un banco de sangre. Se encuentra bajo la responsabilidad de la unidad hospitalaria en la que se encuentre dicho servicio.
3.1.156 Solución aditiva: compuesto nutritivo formulado específicamente para mantener las propiedades benéficas de los productos sanguíneos que contienen eritrocitos y que agregado a éstos incrementan su periodo de vigencia durante su almacenamiento
3.1.157 Solución coloide: suspensión acuosa de proteínas o polisacáridos; el plasma se considera una solución coloide.
3.1.158 Solución crioprotectora: compuesto que impide el daño a las células sanguíneas cuando son sometidas a una temperatura de congelación (-70º C) o ultracongelación (-150º C).
3.1.159 Solución cristaloide: es una solución acuosa de sales minerales y otras moléculas pequeñas solubles en agua. La mayoría de las soluciones cristaloides disponibles comercialmente son isotónicas comparadas para el plasma humano. Estos líquidos se aproximan a las concentraciones de diversos solutos que se encuentran en el plasma y no ejercen un efecto osmótico in vivo.
3.1.160 Título: es la mayor dilución de una muestra de suero o plasma en la que se presenta una reacción considerada como reactiva o positiva.
3.1.161 Transfusión: procedimiento a través del cual se suministra sangre o cualquiera de sus componentes a un ser humano, solamente con fines terapéuticos.
3.1.162 Transfusión ambulatoria: la aplicación de sangre o productos sanguíneos que se efectúa en receptores no hospitalizados.
3.1.163 Transfusión de urgencia: se considera como tal aquélla que cuando un retraso en su aplicación pone en peligro la vida del paciente.
3.1.164 Transfusión masiva: aplicación a un receptor de una cantidad de sangre o productos sanguíneos aproximadamente igual o mayor a su volumen sanguíneo en un lapso de 24 horas. Se considerará como tal la exsanguineotransfusión.
3.1.165 Trastorno de abuso de alcohol: síndrome de dependencia o adicción al alcohol etílico.
3.1.166 Trazabilidad: la capacidad de efectuar el seguimiento de cada unidad de sangre o componente sanguíneo desde el donador hasta su uso terapéutico, fraccionamiento en hemoderivados o su destino final incluyendo su desecho o almacenamiento en serotecas y viceversa.
3.1.167 Triatómino: subfamilia de insectos que se alimentan con sangre de vertebrados, pertenecen a la familia Reduviidae del orden Heteroptera/Hemiptera. Todas las especies son vectores potenciales de la enfermedad de Chagas.
3.1.168 Toxemia gravídica: se le conoce como preeclampsia, una complicación médica que surge durante el embarazo (después de la semana 20) y se caracteriza por el desarrollo de hipertensión, y la presencia de proteínas en la orina.
3.1.169 Unidad de sangre: volumen de sangre (450 mL ±) o componente sanguíneo obtenido para uso terapéutico, de un solo donador, en una sesión de extracción, en una bolsa o recipiente que contenga una solución con propiedades anticoagulantes y conservadoras, adecuadas, suficientes, estériles y carente de pirógenos.
3.1.170 Urgencia transfusional: circunstancia de apremio bajo la cual la no aplicación inmediata de sangre o de productos sanguíneos puede poner en peligro la vida del receptor.
3.1.171 Uso alogénico: cuando el donador y el receptor de la sangre o productos sanguíneos son de la misma especie, aunque no genéticamente idénticos.
3.1.172 Uso autólogo: cuando el donador de sangre o productos sanguíneos es la misma persona que el receptor.
3.1.173 Uso singénico: cuando el donador y el receptor de sangre y productos sanguíneos, son gemelos monocigóticos.
3.1.174 Validación: es una parte del aseguramiento del sistema de calidad que evalúa anticipadamente los pasos involucrados en los procedimientos operativos o de la preparación del producto para asegurar la calidad, efectividad y confiabilidad.
3.1.175 Verificación: la constatación ocular o comprobación mediante muestreo, medición, pruebas de laboratorio, o examen de documentos que se realizan para evaluar la conformidad en un momento determinado.
3.1.176 Valor de corte: cifra que permite diferenciar los resultados reactivos de los no reactivos de una prueba o procedimiento.
3.1.177 Volumen eritrocítico: porción de la sangre circulante formada por la masa total de los glóbulos rojos.
3.1.178 Volumen sanguíneo: porción del cuerpo contenida en el espacio intravascular constituida por el tejido hemático.
3.1.179 Xenotrasplante: procedimiento terapéutico consistente en la transferencia a una persona de órganos, tejidos o células obtenidos de un organismo de distinta especie.
3.2 Para los efectos de este proyecto de Norma, se aplicará la terminología siguiente:
3.2.1 Los términos eritroaféresis, plaquetaféresis, plasmaféresis y granulocitoféresis, se refieren a los procedimientos de aféresis por los cuales los cuales se colectan selectivamente eritrocitos, plaquetas, plasma y granulocitos, respectivamente.
3.2.2 Son procedimientos de transfusión autóloga de reposición inmediata la hemodilución aguda preoperatoria y la recuperación sanguínea.
3.2.3 Se entenderá como equipo, material o proceso crítico, a aquellos que pueden afectar a la calidad del producto o servicio.
3.2.4 Evento, práctica o actividad de riesgo, es aquélla en la que ocurre contacto o traspaso de sangre, secreciones sexuales u otros líquidos corporales de personas que pudieran tener infecciones transmisibles, con sitios del cuerpo de otra persona a través de los cuales el agente infeccioso pudiese penetrar.
4. Símbolos y términos abreviados
4.1 Los símbolos, signos y abreviaturas utilizados en este proyecto de Norma se señalan en la Tabla 2, a continuación:
Tabla 2
Símbolos, signos y abreviaturas
Símbolos o signos |
|||||
% Por ciento |
> Mayor que |
||||
º C Grados Celsius × Signo de multiplicación ± Más o menos |
≤ Igual o menor que ≥ Igual o mayor que ~ Aproximadamente |
||||
< Menor que |
|||||
Abreviaturas (unidades de medida) |
|||||
L |
Litro |
kg |
Kilogramo |
||
g |
Gramo |
mg |
Miligramo |
||
dL |
Decilitro |
Mm Hg |
Milímetros de mercurio |
||
mL |
Mililitro |
mOsm |
Miliosmoles |
||
µL |
Microlitro |
pH |
Potencial de hidrógeno |
||
m |
Metro |
UI |
Unidades internacionales |
||
mm |
Milímetro |
cGy |
Centigray |
||
µm |
Micra, micrón o micrómetro (la millonésima parte de un metro, 1*10-6). |
rpm |
Revoluciones por minuto |
||
Otras abreviaturas |
|||||
BCG |
Bacilo de Calmette y Guerin |
IgG |
Inmunoglobulina G |
||
Cs 137 |
Isótopo de cesio 137 |
IgM |
Inmunoglobulina M |
||
Co 60 |
Isótopo de Cobalto 60 |
LGS |
Ley General de Salud |
||
CPD |
Solución anticoagulante y conservadora con Citrato, Fosfato y Dextrosa |
NAT |
"Nucleid Acid Test" (por sus siglas en inglés) Detección de ácidos nucleícos |
||
CPDA |
Solución anticoagulante y conservadora con Citrato, Fosfato, Dextrosa y Adenina |
OIEA |
Organismo Internacional de Energía Atómica |
||
CPr |
Células progenitoras |
REGLAMENTO |
Reglamento de la Ley General de Salud en Materia de Control Sanitario de la Disposición de Órganos, Tejidos, Células y Cadáveres de Seres Humanos |
||
RPR |
"Rapid Plasm Reagine" (por sus siglas en inglés) Prueba rápida de reaginas |
||||
SECRETARÍA |
Secretaría de Salud |
||||
Factor VIIIc |
Factor VIII coagulante Factor VIII coagulante |
SANGRHE |
Sistema Analítico Nacional para Gestionar Reportes de Hemocomponentes |
||
FEUM |
Farmacopea de los Estados Unidos Mexicanos |
VIH |
Virus de la inmunodeficiencia humana |
||
HTLV I y II |
"Human Leukocytes Antigens". (por sus siglas en inglés) Antígenos Leucocitarios Humanos |
VDRL "Veneral diseases research laboratory"; (por sus siglas en inglés). Prueba no treponémica de antígeno de cardiolipina |
|||
HLA |
"Human Leukocytes Antigens". (por sus siglas en inglés) Antígenos Leucocitarios Humanos |
||||
HPA |
"Human Platelet Antigens". (por sus siglas en inglés) Antígenos Plaquetarios Humanos |
||||
5 Disposiciones generales
5.1 Este proyecto de Norma regulará las actividades relativas a la disposición de sangre y productos sanguíneos con fines transfusionales con el objetivo de incrementar la autosuficiencia de los productos sanguíneos y de garantizar la máxima reducción de los riesgos asociados, promoviendo condiciones uniformes en la terapia transfusional en los establecimientos de prestación de servicios de atención médica del Sistema Nacional de Salud.
5.2 La sangre y productos sanguíneos para uso terapéutico deberán reunir los requisitos de calidad necesarios a fin de que resulten seguros, viables y funcionales. Para ello, la evaluación del donador, la obtención, la extracción, los análisis, conservación, preparación, suministro, transportación, recepción, utilización y, en su caso, destino final se efectuará observando los lineamientos que establece este proyecto de Norma y demás disposiciones aplicables.
5.3 Los establecimientos como son los Centros de Colecta, Centros de Procesamiento de Sangre, Centros de Distribución de Sangre y Productos sanguíneos y Centros de Calificación Biológica funcionan bajo la responsabilidad de un Banco de Sangre autorizado del cual dependen.
5.4 Todos los Servicios de Sangre deberán establecer convenios con los establecimientos con los que realicen disposición de sangre y sus componentes, excepto con los servicios que dependan del propio Banco de Sangre.
5.4.1. En urgencias transfusionales, los Bancos de Sangre o los Servicios de Transfusión Hospitalarios podrán suministrar las unidades de sangre o productos sanguíneos que tuviesen disponibles a otros establecimientos aun sin que medie convenio alguno, siempre y cuando el responsable sanitario del Banco de Sangre, del Servicio de Transfusión Hospitalario o, en su caso, el director de la unidad hospitalaria que hace el envío lo autorice y registre por escrito la eventualidad, sin perjuicio de que con posterioridad se formalice un convenio.
5.5 Los responsables sanitarios deberán laborar conforme al horario de funcionamiento del establecimiento declarado en su Aviso de Responsable Sanitario y podrán asumir máximo hasta dos responsivas, siempre y cuando exista una compatibilidad de horarios. El responsable sanitario de cualquier Servicio de Sangre deberá ser profesional de la salud, con título legalmente expedido y registrado por las autoridades educativas competentes, quien deberá contar con aviso a la Secretaría, deberá acreditar que poseen experiencia no menor de tres años en las actividades inherentes a los establecimientos.
5.6 El responsable sanitario de un servicio de sangre deberá vigilar que las actividades relativas a la disposición de sangre y productos sanguíneos se lleven a cabo:
5.6.1 De conformidad con las disposiciones de este proyecto de Norma y demás disposiciones aplicables;
5.6.2 En condiciones de máxima seguridad, bienestar y respeto para los donadores, los receptores, el personal de salud, voluntarios y visitantes. Estas condiciones deberán mantenerse dentro de los establecimientos, así como, en las colectas externas que lleve a cabo los servicios de sangre, y
5.6.3 En apego a los lineamientos y principios internacionales de buenas prácticas de fabricación para productos sanguíneos (consúltese el documento referenciado en el inciso 22.6 de este proyecto de Norma), y
5.6.4 La observancia y aplicación del sistema de gestión de la calidad para supervisar su cumplimiento.
5.7 Para garantizar la seguridad y calidad de las unidades de sangre y productos sanguíneos, así como, la de los servicios prestados, los Servicios de Sangre, deberán contar con un sistema de gestión de la calidad, mismo que será documentado, el cual deberá incluir procedimientos normalizados de operación, guías e instructivos claros, comprensibles que describan todos los procedimientos técnicos y administrativos que realiza el establecimiento, los cuales se deberán mantener, actualizar y permanecer accesibles al personal.
5.8 El personal que labora en los Servicios de Sangre deberá conocer, implementar, mantener y observar lo dispuesto en los documentos que integran el Sistema de Gestión de la Calidad.
5.9 Todas las actividades relativas a la disposición de sangre y productos sanguíneos deberán registrarse, de forma que permitan garantizar la trazabilidad de las unidades, desde su extracción hasta su uso terapéutico o destino final y viceversa. Para efectos de ese proyecto de Norma, una actividad no registrada se considerará como no efectuada.
5.10 Operatividad, procesos e interrelaciones de los Servicios de Sangre:
5.10.1 Banco de Sangre.
5.10.1.1 Los Bancos de Sangre, deberán realizar, los procedimientos y actividades para la promoción y fomento de la donación voluntaria y altruista, atención de los candidatos a donar, obtención de sangre total, obtención de productos sanguíneos, a través de procedimientos de aféresis, tamizaje para marcadores de serología infecciosa, pruebas suplementarias y confirmatorias para agentes trasmisibles por transfusión, pruebas de inmunohematología, separación de productos sanguíneos, conservación de unidades, destino final de unidades, control de calidad de productos sanguíneos y hemovigilancia del proceso de donación, garantizando la trazabilidad de las unidades, desde su extracción hasta uso terapéutico o destino final. Desde el punto de vista operativo también tendrá a cargo el proceso de capacitación, evaluación continua y actualización del personal operativo que labore en su establecimiento. Todos los aspectos técnico-administrativos deberán basarse en un Sistema de Gestión de Calidad, de conformidad con los incisos 5.8 y 5.11 de este proyecto de Norma.
5.10.1.2 En caso de que se considere necesario y cuente con la infraestructura suficiente, podrá realizar procedimientos especiales como irradiación de productos sanguíneos, leucodepleción durante el procesamiento, inactivación de patógenos, realización de mezcla de productos sanguíneos o fracciones pediátricas, entre otros procedimientos. Así mismo, podrá realizar colectas externas de sangre y productos sanguíneos siempre que se garantice la calidad y seguridad en todo el proceso de disposición de sangre y sus componentes.
5.10.1.3 Si el Banco de Sangre se encuentra dentro de una unidad hospitalaria, estará bajo su responsabilidad la aplicación de la sangre y sus productos, así como la hemovigilancia del proceso de transfusión incluyendo la identificación, reporte, notificación y manejo de las reacciones adversas a la transfusión y deberá de contar con un Comité de Medicina Transfusional, el cual tendrá que notificar instauración, ratificación de manera anual y funciones al Centro Nacional de la Transfusión Sanguínea, preferentemente tomando como base las Recomendaciones para la Conformación, Estructura y Funcionamiento del Comité de Medicina Transfusional en los Servicios de Salud.
5.10.1.4. El banco de sangre podrá recibir sangre total y productos sanguíneos, en los términos que establezcan en un convenio, de los siguientes establecimientos:
5.10.1.4.1 Centros de Colecta;
5.10.1.4.2 Centros de Distribución de Sangre y Componentes Sanguíneos;
5.10.1.4.3 Otros bancos de sangre, y
5.10.1.4.4 Servicios de Transfusión Hospitalarios.
5.10.1.5 El Banco de Sangre podrá enviar productos sanguíneos bajo el actuar de un convenio a:
5.10.1.5.1 Servicios de Transfusión Hospitalarios;
5.10.1.5.2 Centros de Distribución de Sangre y Productos sanguíneos, y
5.10.1.5.3 Bancos de Sangre.
5.10.1.6 Contarán con un responsable sanitario, quien deberá vigilar el cumplimiento de las disposiciones de este apartado en los servicios de sangre que tuviese a su cargo.
5.10.2 Centro de Procesamiento de Sangre.
5.10.2.1 Los Centros de Procesamiento de Sangre deberán actuar bajo la responsabilidad de un Banco de Sangre, que cuente con licencia sanitaria vigente; deberán realizar la separación de productos sanguíneos, liberación de sangre segura, conservación, destino final de unidades y control de calidad de insumos, reactivos y productos sanguíneos. Desde el punto de vista operativo, también, tendrán a cargo el proceso de capacitación, evaluación continua y actualización del personal operativo que labore en su establecimiento. Todos los aspectos técnico-administrativos deberán basarse en un Sistema de Gestión de la Calidad en conformidad con lo establecido en los incisos 5.8 y 5.11 de este proyecto de Norma.
5.10.2.2 El Centro de Procesamiento, en caso de ser necesario y cuente con la infraestructura suficiente, podrá realizar procedimientos especiales como irradiación de productos sanguíneos, leucodepleción durante el procesamiento, inactivación de patógenos, realización de mezcla de productos sanguíneos o fracciones pediátricas, entre otros procedimientos, deberá contar con el equipamiento y mantenimiento de equipos e instrumentos, así como calibraciones respectivas cuando apliquen.
5.10.2.3 El Centro de Procesamiento podrá recibir sangre total y productos sanguíneos para su procesamiento, bajo el actuar de un convenio de los Centros de Colecta.
5.10.2.4 El Centro de Procesamiento, podrá enviar productos sanguíneos bajo el actuar de un convenio a:
5.10.2.4.1 Centros de Distribución de Sangre y Productos sanguíneos, y
5.10.2.4.2 Centro de Calificación Biológica (muestras de sangre).
5.10.2.5 Centro de Procesamiento, contará con un responsable sanitario, quien deberá vigilar el cumplimiento de las disposiciones de esta Norma.
5.10.3 Centro de colecta.
Los Centros de Colecta actuarán bajo la responsabilidad de un Banco de Sangre autorizado. Deberán realizar los procedimientos y actividades para la promoción y fomento de la donación voluntaria y altruista, atención y valoración (del médico correspondiente), de los candidatos a donar, obtención de sangre total, obtención de productos sanguíneos, a través de procedimientos de aféresis, pruebas de inmunohematología básica para donadores, conservación y embalaje de unidades, destino final de unidades y hemovigilancia del proceso de donación. También podrá realizar colectas externas.
Desde el punto de vista operativo, también, tendrán a cargo el proceso de capacitación, evaluación continua y actualización del personal operativo que labore en su establecimiento. Todos los aspectos técnico-administrativos deberán basarse en un Sistema de Gestión de la Calidad, de conformidad con lo establecido en los incisos 5.8 y 5.11 de este proyecto Norma.
Se le permitirá el procesamiento de sangre total, conforme a los dispuesto en el capítulo 8 denominado "Extracción de unidades de sangre y productos sanguíneos para uso alogénico" de este proyecto de Norma, cuando la geografía y/o accesibilidad no permita el traslado de la sangre total en menos de 4 horas hasta el sitio donde se llevará a cabo su procesamiento.
5.10.3.1 El Centro de Colecta, podrá enviar sangre total, componentes y muestras sanguíneos bajo el actuar de un convenio para su procesamiento y análisis a:
5.10.3.1.1 Bancos de Sangre;
5.10.3.1.2 Centro de Procesamiento de Sangre, y
5.10.3.1.3 Centro de Calificación Biológica (muestras de sangre exclusivamente).
5.10.3.2 El Centro de Colecta deberá contar con un responsable sanitario, el cual, deberá vigilar el cumplimiento de las disposiciones de esta Norma, y deberá estar capacitado en el manejo de reacciones o efectos adversos asociadas a la donación.
El responsable sanitario de un Banco de Sangre que tengan bajo su responsabilidad centros de colecta, deberá supervisar que éstos tengan un sistema de gestión de calidad aprobado o la observancia del establecido por el propio Banco de Sangre del cual depende el Centro de Colecta.
5.10.4 Centro de Distribución de Sangre y Productos sanguíneos.
Los Centros de Distribución de Sangre y Productos sanguíneos funcionarán bajo la responsabilidad de un Banco de Sangre autorizado y deberán realizar la recepción, conservación, gestión y distribución de unidades de productos sanguíneos a otros establecimientos, así como su destino final cuando aplique. Desde el punto de vista operativo también tendrá a cargo el proceso de capacitación, evaluación continua y actualización del personal operativo que labore en su establecimiento. Todos los aspectos técnico-administrativos deberán basarse en un Sistema de Gestión de la Calidad, de conformidad con lo establecido en los incisos 5.8 y 5.11 de este proyecto de Norma.
5.10.4.1 El Centro de Distribución de Sangre y Productos sanguíneos podrá recibir productos sanguíneos bajo el actuar de un convenio de los siguientes de establecimientos:
5.10.4.1.1 Bancos de Sangre;
5.10.4.1.2 Centros de Procesamiento de Sangre, y
5.10.4.1.3 Servicios de Transfusión Hospitalarios.
5.10.4.2 El Centro de Distribución de Sangre y Productos sanguíneos también podrá enviar productos sanguíneos bajo el actuar de un convenio a:
5.10.4.2.1 Bancos de Sangre, y
5.10.4.2.2 Servicios de Transfusión Hospitalarios.
5.10.4.3 El Centro de Distribución de Sangre y Productos sanguíneos deberá contar con un responsable sanitario, el cual deberá vigilar el cumplimiento de las disposiciones de este Proyecto de Norma.
5.10.5 Servicio de Transfusión Hospitalario.
Los Servicios de Transfusión Hospitalarios, deberán realizar los procedimientos y actividades para la promoción y fomento de la donación voluntaria y altruista, pruebas de inmunohematología (grupos sanguíneos, pruebas cruzadas, rastreo de anticuerpos irregulares), conservación de unidades, destino final de unidades, aplicación de productos sanguíneos en los servicios hospitalarios con fines terapéuticos y hemovigilancia del proceso de transfusión, incluyendo la identificación, reporte, notificación y manejo de las reacciones adversas a la transfusión. Desde el punto de vista operativo, también tendrá el proceso de capacitación, evaluación continua y actualización del personal operativo que labore en su establecimiento. Todos los aspectos técnico-administrativos deberán basarse en un Sistema de Gestión de la Calidad.
Deberá de contar con un Comité de Medicina Transfusional, el cual tendrá que notificar instauración, ratificación de manera anual y funciones al Centro Nacional de la Transfusión Sanguínea, preferentemente tomando como base las Recomendaciones para la Conformación, Estructura y Funcionamiento del Comité de Medicina Transfusional en los Servicios de Salud, disponibles en el sitio: https://www.gob.mx/cnts/documentos/recursos-guias-y-documentos-en-medicina-transfusional
En caso de que se considere necesario y cuente con la infraestructura suficiente, podrá realizar procedimientos especiales como irradiación de productos sanguíneos (para lo cual deberá contar con la licencia aplicable en la materia), procesamiento de leucodepleción, realización de mezcla de productos sanguíneos o fracciones pediátricas, entre otros procedimientos.
5.10.5.1 El Servicio de Transfusión Hospitalario podrá recibir productos sanguíneos bajo el actuar de un convenio de:
5.10.5.1.1 Bancos de sangre, y
5.10.5.1.2 Centros de Distribución de Sangre y Productos sanguíneos.
5.10.5.2 El Servicio de Transfusión Hospitalario podrá enviar productos sanguíneos bajo el actuar de un convenio a:
5.10.5.2.1 Servicios de Transfusión hospitalarios (en caso de alguna urgencia acreditada);
5.10.5.2.2 Centros de Distribución de Sangre y Productos sanguíneos, y
5.10.5.2.3 Bancos de Sangre
5.10.5.3 El Servicio de Transfusión contará con un responsable sanitario del servicio, experiencia y conocimientos comprobables en medicina transfusional de al menos 3 años.
5.10.6 Centro de Calificación Biológica.
Los Centros de Calificación Biológica funcionarán bajo la responsabilidad de un Banco de Sangre podrán realizar los procesos de tamizaje para marcadores de serología infecciosa, pruebas suplementarias y confirmatorias para agentes trasmisibles por transfusión, pruebas de amplificación de ácidos nucleicos aplicadas a servicios de sangre, así como pruebas de inmunohematología. Deberán recibir exclusivamente muestras sanguíneas, no tienen las atribuciones de recepción, conservación o distribución de sangre y sus productos sanguíneos. Todos los aspectos técnico-administrativos deberán basarse en un Sistema de Gestión de la Calidad, de conformidad con este proyecto de Norma, Para la Disposición de Sangre Humana y sus Componente con Fines Terapéuticos.
Desde el punto de vista operativo también tendrá a cargo el proceso de capacitación, evaluación continua y actualización del personal operativo que labore en su establecimiento. Todos los aspectos técnico-administrativos deberán basarse en un Sistema de Gestión de la Calidad, de conformidad con lo establecido en los incisos 5.8 y 5.11 de este proyecto de Norma.
5.10.6.1 El Centro de Calificación Biológica podrá recibir muestras sanguíneas para análisis de los siguientes establecimientos:
5.10.6.1.1 Centros de Colecta;
5.10.6.1.2 Bancos de Sangre;
5.10.6.1.3 Centros de Procesamiento de Sangre, y
5.10.6.1.4 Servicios de Transfusión Hospitalarios (pruebas de inmunohematología y de estudio de reacciones adversas a la transfusión).
5.10.6.2 El Centro de Calificación Biológica deberá contar con un responsable sanitario el cual, deberá vigilar el cumplimiento de las disposiciones de este Proyecto de Norma.
5.10.7 Las actividades de los Bancos de Sangre y de los Servicios de Transfusión Hospitalarios podrán llevarse a cabo directamente en estos establecimientos o a través de otros Bancos de Sangre o Servicios de Transfusión Hospitalario, siempre y cuando cumplan con los requisitos establecidos en este proyecto de Norma y en las disposiciones que resulten aplicables con el fin de garantizar la seguridad sanguínea, pero en cualquier caso serán responsables solidarios de dichas actividades.
5.10.8 El responsable sanitario de un servicio de sangre, que funja como tal en un establecimiento de servicio de sangre podrá asumir máximo hasta dos responsivas, siempre y cuando exista una compatibilidad de horarios, en los que se desempeñe en cada uno de los servicios.
5.11 Sistema de Gestión de la Calidad en los Servicios de Sangre.
Los responsables sanitarios de los Servicios de Sangre deberán implementar el sistema de gestión de la calidad a que se refiere el inciso 5.8 de este Proyecto de Norma, mediante un Manual de Calidad, para lo cual podrá auxiliarse de un profesional responsable del Sistema de Gestión de la Calidad, este asumirá las mismas responsabilidades enfocadas a dirigir y controlar la organización relativa al manejo documental y deberá cumplir con lo siguiente:
5.11.1 Abarcará la estructura de la organización y tendrá la descripción de todas las actividades individuales y colectivas;
5.11.2 Incluirá las políticas y objetivos de calidad, planificación, procesos, procedimientos, verificación, control, control de cambios, aseguramiento y mejora continua de la calidad de las actividades que realiza el establecimiento y los recursos necesarios para su desarrollo, mismos que estarán asentados en el sistema documental y en los procedimientos normalizados de operación;
5.11.3 Estará continuamente actualizados mediante revisiones conjuntas, de periodicidad programada, efectuadas cuando menos una vez al año, o bien, cuando resulte necesario;
5.11.4 El sistema documental y los procedimientos normalizados de operación sean técnicos o administrativos, así como, sus modificaciones o adecuaciones que resulten necesarias deberán ser aprobados por el responsable sanitario y quedarán debidamente registradas:
5.11.5 Evidencia de que todos los equipos cuenten con certificados de calificación, así como, de que se ha efectuado su calibración, verificación, monitoreo, mantenimiento preventivo y correctivo y capacitación del personal para el uso adecuado de los mismos;
5.11.6 Implementará el uso de herramientas estadísticas que permitan realizar el monitoreo de todo el proceso y mostrará evidencia de su implementación, que apoye su adecuado funcionamiento, permite analizar áreas de mejora continua, atendiendo a las desviaciones encontradas;
5.11.7 Asegurarse que el sistema documental requerido por el sistema de gestión de la calidad tenga un lenguaje técnico, inequívoco pero comprensible, esté disponible y adecuado para su uso, dónde y cuándo se necesite, esté protegido adecuadamente (por ejemplo, de las modificaciones no intencionadas o el acceso por personal sin autoridad para consultar la información), y
5.11.8 Establecer anualmente las metas de colecta de sangre y productos sanguíneos que, permitan satisfacer las demandas transfusionales necesarias para mantener la continuidad de los procesos de atención médica que requieren transfusión sanguínea, las demandas transfusionales deberán contener una lógica numérica basada en la evidencia científica disponible, con el fin de incrementar la seguridad transfusional y disminuir el desecho de sangre y productos sanguíneos por caducidad.
Para la estructuración de todos los documentos incluidos en el Sistema de Gestión de Calidad, se deberá asegurar que la información documentada se encuentre: disponible para su uso, protegida adecuadamente y clasificada de conformidad con el procedimiento de control de documentos.
5.12 El responsable sanitario del establecimiento, el o los encargados del sistema de gestión de la calidad deberán observar las disposiciones siguientes:
5.12.1 Dirigir y controlar la organización con respecto a la calidad para conducir a la misma hacia la mejora de su desempeño;
5.12.2 Establecer y promover las políticas y objetivos de la calidad de la organización;
5.12.3 Asegurarse de la disponibilidad de los recursos necesarios para el mantenimiento del sistema de gestión de la calidad;
5.12.4 Participar con el resto del personal para alcanzar y mantener la eficacia del sistema de gestión de la calidad, así como en su mejora continua, con base en la identificación, planificación, desarrollo, implementación y evaluación de procesos que permitan mejorarlo;
5.12.5 Asegurarse de que los procesos y los procedimientos operativos específicos que se emplean son acordes con el sistema de gestión de la calidad y que se encuentren disponibles en su lugar de uso;
5.12.6 Establecer los procedimientos para la identificación, obtención de datos, análisis, diseño, desarrollo y seguimiento de las acciones correctivas y preventivas para mejorar y actualizar los procesos;
5.12.7 Asegurarse de que los documentos obsoletos estén adecuadamente identificados y que no estén en uso;
5.12.8 Asegurar la confidencialidad y custodia de los documentos que lo requieran, de acuerdo con la legislación aplicable;
5.12.9 Asegurarse de que se identifican los documentos externos y que se controla adecuadamente su distribución, y
5.12.10 Asegurarse de que el personal sea formado en el uso y la aplicación de los procedimientos normalizados de operación que integran el sistema de gestión de la calidad y a los demás documentos a que haga referencia el sistema, así como, a los requisitos para su desarrollo.
5.13 Los Bancos de Sangre, Centros de Colecta y los Servicios de Transfusión Hospitalarios, deberán implementar programas de educación, información, sensibilización y reclutamiento en la comunidad dentro de su área de influencia para fomentar la donación voluntaria y altruista, periódica y responsable con la finalidad de mantener una fuente de donadores sanos y comprometidos.
5.14 Toda donación de sangre o productos sanguíneos deberá ser voluntaria, libre de coacción y no remunerada. No deberá otorgarse al donador pago alguno, tanto en dinero en efectivo ni en formas equivalentes. No se considerarán como pago el refrigerio que se les da después de la donación, el pago de los costos estrictamente necesarios para el traslado al sitio de la donación o pequeños obsequios tales como bolígrafos, prendedores promocionales y otros artículos semejantes.
5.15 Los establecimientos de los Servicios de Sangre, de acuerdo con las actividades que realizan, deberán contar con lo siguiente:
5.15.1 Un diseño arquitectónico acorde con las funciones que desempeña el establecimiento y que permita:
5.15.1.1 Que las actividades se desarrollen de manera eficaz y con mínimos riesgos para la salud del personal, de los donantes, los receptores, voluntarios y visitantes;
5.15.1.2 Que exista privacidad para los donadores y, en su caso, los receptores;
5.15.1.3 Que haya un control adecuado para el acceso a las áreas restringidas, y
5.15.1.4 Que facilite el orden y limpieza de las áreas de trabajo.
5.15.2 Equipos, instrumentos, reactivos, materiales e insumos necesarios para el desarrollo de las funciones para las cuales el establecimiento está autorizado.
5.15.3 Personal suficiente de acuerdo con las características del servicio de sangre que se trate e idóneo a través de su perfil profesional y formado para el desempeño de sus actividades dentro del mismo.
5.15.4 Documentación y registros de las actividades relativas a la disposición de sangre y productos sanguíneos, de conformidad con lo dispuesto en el capítulo 20 denominado "Procedimientos normalizados de operación, guías, instructivos, documentos y registros" de este proyecto de Norma.
5.15.5 Las demás que señala el Reglamento en materia.
5.16 Funciones generales del responsable sanitario de un Servicio de Sangre.
5.16.1 Asignará las responsabilidades y funciones a cada trabajador, asegurándose de que queden bien definidas, documentadas y que sean del conocimiento del resto del personal.
5.16.2 Se asegurará que el personal reciba la formación necesaria y apropiada para la realización de su trabajo y que se mantenga continuamente capacitado y actualizado.
5.16.3 Antes de que un miembro del personal acceda a la realización de un puesto determinado, deberá ser evaluado y quedará documentado que la formación, los conocimientos y la experiencia concuerdan con las exigencias requeridas para el cargo.
5.16.4 Establecerá evaluaciones periódicas de la capacitación y el desempeño del personal, así como de la eficacia de los programas de capacitación y actualización en materia de disposición de sangre y productos sanguíneos de conformidad con los avances científicos y tecnológicos. Asimismo, se asegurará de que se disponga de registros detallados sobre las acciones a que se refiere este apartado.
5.16.5 Establecerá los procedimientos que aseguren la protección de la información confidencial, de conformidad con las disposiciones aplicables en materia de Transparencia, Acceso a la Información y Protección de Datos Personales.
5.16.6 Se asegurará de que se establezcan los procesos de comunicación apropiados dentro del establecimiento y, en su caso, con los centros de colecta que tuviesen, con la unidad hospitalaria donde se ubica el establecimiento o con los servicios de sangre que tengan algún mecanismo de convenio con el servicio de sangre bajo su responsabilidad.
5.16.7 Informará y capacitara periódicamente al personal operativo adscrito al servicio de sangre de su responsabilidad, acerca de la normatividad aplicable al establecimiento, los documentos normativos que emita la Secretaría de Salud en materia de uso y disposición de sangre y; atenderá las capacitaciones y avisos que la Secretaría de Salud a través del Centro Nacional de la Transfusión Sanguínea emita en lo relativo a este proyecto de Norma.
5.17 El Banco de Sangre y Servicio de Transfusión, deberán disponer de un profesional de la salud, responsable de gestionar los procesos de hemovigilancia que posibiliten la detección, registro, análisis de la información y notificación de los incidentes y de las reacciones o efectos adversos e inesperados de la donación o de la transfusión.
5.18 Los documentos empleados durante el proceso de registro deberán incluir información adecuada y suficiente de cualquier evento, reacción, efecto adverso que pudiera producirse o detectarse durante o después de la donación o la transfusión.
5.19 Ante eventos, complicaciones o efectos adversos e inesperados de la donación o transfusión, los Bancos de Sangre, Servicios de Transfusión Hospitalarios y los establecimientos de atención médica, deberán implementar medidas de carácter preventivo o correctivo.
5.20 Las medidas correctivas o preventivas adoptadas deberán registrarse y se llevará a cabo regularmente un seguimiento y análisis de su grado de cumplimiento y eficacia.
5.21 La notificación se hará al Centro Nacional de la Transfusión Sanguínea, de conformidad con los formatos que para ello establezca o a través de a través de la plataforma digital referida en el inciso 19.2 de este Proyecto de Norma y al comité de medicina transfusional que el establecimiento tuviese.
6 Información, consentimientos y atención para donantes y receptores
6.1 Todo material informativo, notificación, carta de consentimiento informado o cualquier documento relativo a las actividades de la disposición de sangre y productos sanguíneos, que se proporcione a un donante, a un receptor o, en su caso, al padre, la madre, el tutor, quien ejerza la patria potestad o el representante legal, deberá estar escrito en español, de manera clara y completa. En caso de que la persona no entienda el español, la información le será proporcionada en su propio idioma o lengua; de no ser esto posible, se transmitirá de manera verbal auxiliado por un intérprete. En cualquiera de los casos, habrá una versión escrita en español y, en su caso, en el idioma o lengua que se hable en la región, así como el acceso a un Intérprete de Lengua de Señas Mexicana, de ser requerido. De manera optativa se podrá usar una herramienta tecnológica en el idioma o lengua que se hable en la región o de Lengua de Señas Mexicana.
6.2 Información a los donantes de sangre y de productos sanguíneos.
6.2.1 Antes de cada donación de sangre o productos sanguíneos, los Bancos de Sangre y Centros de Colecta deberán proporcionar, a través de la asistente social y/o trabajadora social, a los candidatos a donar sangre o productos sanguíneos para uso alogénico o autólogo, de manera oral y escrita, quedando constancia de ello, material educativo e informativo, preciso y en lenguaje comprensible, acerca de lo siguiente:
6.2.1.1 De los requisitos generales de salud de los donantes, estilos de vida saludables y sobre los beneficios terapéuticos para el receptor.
6.2.1.2 Se deberá de proporcionar en el material informativo al que se hace referencia en el inciso 6.2.1 de este Proyecto de Norma, los teléfonos y medios de contacto para cualquier notificación que requieran hacer los donantes posterior a la donación.
6.2.1.3 Los eventos, actividades y prácticas sexuales de riesgo que excluyen temporal o definitivamente de la donación, por suponer un riesgo de infección por agentes transmisibles por transfusión, en especial el síndrome de la inmunodeficiencia humana y las hepatitis virales, así como sobre la importancia de no donar sangre si les es aplicable alguna de ellas (véase inciso 3.2.4 de este proyecto de Norma). Asimismo, se les informará sobre las circunstancias que contraindican la donación por representar un riesgo para su propia salud.
6.2.1.4 Sobre la influencia favorable en la seguridad transfusional de la donación voluntaria y altruista de repetición, así como sobre la conveniencia para la sociedad, el país, para los receptores y para el mismo donante de establecer un compromiso de donar sangre de manera regular y programada.
6.2.1.5 Que a quienes accedan a ser donantes de repetición se les invitará a futuras donaciones, de acuerdo con las necesidades sanguíneas y respetando los intervalos individuales entre cada donación; asimismo, en su caso, la fechas subsecuentes y lugares donde se instalará una unidad móvil.
6.2.1.6 Sobre el procedimiento de donación habitual y mediante aféresis, su tiempo estimado, el volumen de sangre o del componente sanguíneo que se le extrae, el número de veces que pueden donar en el lapso de un año, las posibles reacciones o efectos adversos que pueden aparecer durante su transcurso y después del mismo, las medidas para solventarlos y los cuidados que deben tener en el periodo que sigue a la donación, así como en el sitio de la venopunción.
6.2.1.7 Sobre la importancia de notificar al Banco de Sangre o, en su caso, al Centro de Colecta, cualquier síntoma, signo o acontecimiento posterior a la donación que pudiera hacer inadecuada la sangre y sus componentes para uso terapéutico.
6.2.1.8 En su caso, sobre cualquier causa de exclusión que el donante detecte y que no hubiera sido tomada en cuenta en alguna donación previa.
6.2.1.9 Sobre el derecho que tienen de hacer preguntas en cualquier momento y de que puede retirarse o excluirse en cualquier fase de la donación.
6.2.1.10 Sobre los análisis previos y posteriores a la donación y que de obtener resultados no aptos por representar riesgos a la salud del donante o del receptor supondrá su exclusión como donante o el destino final de la sangre y productos sanguíneos que se hubieran recolectado.
6.2.1.11 Sobre la obligatoriedad que establece la Ley de notificar a la Secretaría o a la autoridad sanitaria más cercana de las enfermedades transmisibles posteriormente a su diagnóstico o sospecha diagnóstica, en los términos que establece la misma Ley.
6.2.1.12 Que sus datos personales, los referentes a su donación y resultados de las pruebas de laboratorio serán tratados de manera confidencial, de conformidad con las disposiciones aplicables en materia de Transparencia, Acceso a la Información y Protección de Datos Personales. Asimismo, se le explicará la posibilidad de corrección de los datos que serán mantenidos en el Banco de Sangre, Centro de Colecta o Servicio de Transfusión Hospitalario.
6.2.1.13 A los candidatos a procedimientos de transfusión autóloga por depósito previo o, en su caso, al padre, la madre, el tutor, quien ejerza la patria potestad o el representante legal, se les informará además, sobre la posibilidad de su exclusión del procedimiento y las razones médicas por las cuales no debe efectuarse, así como la posibilidad de que la sangre o componentes autólogos puedan ser insuficientes para cubrir las necesidades previstas y por ende la probabilidad de requerirse sangre o productos sanguíneos alogénicos.
6.2.2 El Banco de Sangre, deberá notificar al donante de sangre o productos sanguíneos, para uso alogénico o autólogo, los resultados de los análisis de laboratorio que indican que la donación no es apta. Tratándose de pruebas para la detección de agentes infecciosos transmisibles, la notificación deberá hacerse en un lapso que no exceda de ocho días hábiles contados a partir de obtener un resultado confirmado y con un mínimo de tres intentos de localización.
6.2.3 En caso de donantes regulares o de repetición, que en alguna donación se detecte la presencia de algún marcador de un agente transmisible por transfusión, el Banco de Sangre o Servicio de Transfusión Hospitalario, deberá localizar y notificar al o a los receptores de donaciones previas con el fin de investigar la posibilidad de una transmisión de un agente infeccioso durante el periodo de ventana que el donante estuviese. La notificación deberá hacerse en un lapso que no exceda de ocho días contados a partir de obtener un resultado confirmado y con un mínimo de tres intentos de localización.
6.2.4 Para la notificación de los resultados de los análisis de laboratorio que indican que la donación no es apta, se deberá proceder como sigue:
6.2.4.1 Los resultados se entregarán por escrito por personal médico autorizado (en caso de un resultado anómalo), exclusivamente al interesado. Tratándose de menores o en estado de interdicción sometidos a procedimientos de autotransfusión, los resultados se entregarán a cualquiera de las personas siguientes: el padre, la madre, el tutor, quien ejerza la patria potestad o el representante legal;
6.2.4.2 La entrega de resultados deberá hacerse siempre mediante consejería y de manera personal. Se proporcionará la orientación pertinente, a fin de que puedan acceder a una atención médica oportuna;
6.2.4.3 Todas las acciones realizadas para los fines de este apartado deberán registrarse en el expediente del donante o del paciente, y
6.2.4.4 El interesado o, en su caso, cualquiera de las personas señaladas en el inciso 6.1, acreditarán la recepción de la notificación mediante su firma o huella dactilar.
6.2.5 En caso de que en los análisis de laboratorio se advierta que la donación es idónea, los resultados se entregarán por escrito y/o correo electrónico conforme a lo notificado en el Aviso de Privacidad sólo si el interesado o, en su caso, el padre, la madre, el tutor, quien ejerza la patria potestad o el representante legal así lo solicitan. El resultado de laboratorio anómalo se deberá entregar por escrito por el personal médico del Banco de Sangre.
6.3 Consentimiento informado para donantes de sangre o productos sanguíneos y para los receptores de una transfusión.
6.3.1 Previo al procedimiento los donantes de sangre y productos sanguíneos, los receptores y las personas que vayan a someterse a procedimientos de transfusión autóloga, deberán otorgar su consentimiento escrito, con firma de autorización o, en su caso, con huella dactilar, una vez que hubieran recibido información completa y a su satisfacción sobre el acto de disposición de que se trate, en un documento denominado "carta de consentimiento informado" (véase capítulo 20 denominado "Procedimientos normalizados de operación, guías, instructivos, documentos y registros" de este proyecto de Norma). El formato también debe servir como evidencia de rechazo al procedimiento.
6.3.2 Los otorgantes de una carta consentimiento informado, deberán ser mayores de edad, estar en pleno uso de sus facultades mentales y actuar de manera libre, sin coacción física, moral o económica.
6.3.3 Tratándose de menores o en estado de interdicción que fuesen a recibir una transfusión o someterse a algún procedimiento para uso autólogo, la carta consentimiento informado la otorgará el padre, la madre, el tutor, quien ejerza la patria potestad o el representante legal.
6.3.4 Los responsables de recabar la carta del consentimiento informado son:
6.3.4.1 El responsable sanitario de un Banco de Sangre, Centro de Colecta, o bien el personal médico asignado por éstos, para el caso de donantes de sangre o productos sanguíneos para fines de transfusión alogénica;
6.3.4.2 El responsable sanitario o el médico tratante de un Banco de Sangre o de un Servicio de Transfusión Hospitalario, para efectuar un procedimiento de transfusión autóloga mediante depósito previo;
6.3.4.3 El médico que indique o el que vaya a ejecutar algún procedimiento de hemodilución aguda preoperatoria o de recuperación sanguínea perioperatoria para uso autólogo, y
6.3.4.4 El médico tratante o el médico que indique una transfusión en algún receptor.
6.3.5 En caso de urgencia para aplicar una transfusión en un receptor que no esté en uso pleno de sus facultades mentales o ante la necesidad de efectuar un procedimiento de transfusión autóloga en menores o en estado de interdicción de otorgar su consentimiento y en ausencia de un otorgante facultado para el ejercicio de tal derecho, el médico tratante, llevará a cabo el procedimiento de que se trate, dejando constancia en el expediente clínico.
6.3.6 Tratándose de receptores que requieran continuamente transfusiones, el consentimiento informado se obtendrá la primera vez que sea requerida la transfusión y en las subsecuentes bastará con registrar la conformidad del receptor en su expediente clínico.
6.3.7 El receptor que requiera transfusión de productos sanguíneos, con independencia de su postura religiosa, conciencia, ética e ideológica, tiene derecho a recibir información suficiente, clara, oportuna, y veraz, así como la orientación que sea necesaria respecto de su estado salud y sobre los riesgos, beneficios y alternativas del procedimiento terapéutico que se le indique o aplique, por lo que una vez garantizada la comprensión de la información, los receptores tienen el derecho de aceptar o rechazar una transfusión. El rechazo de esta no lo condiciona a recibir una atención médica fuera de los principios de los derechos humanos o a la negación de atención médica o quirúrgica alguna.
6.4 Refrigerio y suplementos vitamínicos y minerales para los donantes.
6.4.1 Los Bancos de Sangre y Centros de Colecta deberán proporcionar a los donantes:
6.4.1.1 Un refrigerio después de cada donación, con un volumen, entre líquidos y sólidos (evitando los alimentos pre envasados, ultra procesados y con la menor cantidad de sellos) el cual deberá ser similar al volumen de la sangre o componente sanguíneo extraído. De acuerdo con el inciso 4.5.3 de la NOM-051-SCFI/SSA1-2010 y las Guías Alimentarias Saludables y Sostenibles para la Población Mexicana 2023. Además, se deberá contar con un área adecuada para tal fin que, deberá ser distinta al área de sangrado.
6.4.1.2 Cuando se juzgue indicado y de no haber intolerancia, el personal médico del Banco de Sangre o del Centro de Colecta prescribirá suplementos de hierro, folatos o ambos.
6.4.1.3 Un área física donde se otorgue el refrigerio tendrá condiciones adecuadas de acceso, iluminación, ventilación, temperatura y dentro de las instalaciones del Banco de Sangre o Centro de Colecta que permita la atención oportuna en casos de reacción adversa a la donación.
6.4.2 Tratándose de procedimientos de depósito previo para uso autólogo, los Bancos de Sangre y los Servicios de Transfusión Hospitalario, deberán proporcionar a los pacientes lo señalado en el inciso anterior, observando lo siguiente:
6.4.2.1 El alimento será proporcionado siempre que su condición lo permita y mientras no estén sujetos a algún régimen dietético especial, y
6.4.2.2 Cuando se juzgue indicado y de no haber intolerancia se prescribirán suplementos de hierro, folatos o estimulantes de la hematopoyesis. En el caso de los suplementos de hierro, es recomendable iniciar su administración antes de empezar el programa de extracciones y hasta antes de la cirugía.
7 Selección de donantes para uso terapéutico alogénico
7.1 El objetivo del proceso de selección de los candidatos a donar es determinar si la persona se encuentra en condiciones adecuadas para poder realizar la donación sin que existan riesgos para su salud ni para la del futuro receptor.
7.2 El donante deberá cumplir una serie de requisitos mínimos establecidos para poder realizar una donación, en casos de duda prevalecerá el criterio médico el que en todo momento observará las disposiciones éticas y legales aplicables.
7.3 El donante que proporcione su sangre y productos sanguíneos para uso alogénico podrá corresponder a las categorías siguientes:
7.3.1 Voluntario y altruista;
7.3.2 Familiar o de reposición;
7.3.3 Designado;
7.3.4 Dirigido;
7.3.5 Regular, o
7.3.6 De repetición.
Por seguridad transfusional, deberán evitarse las donaciones familiares o de reposición y las donaciones dirigidas.
Los donantes mencionados en los incisos 7.3.1, 7.3.2, 7.3.3 y 7.3.4 podrán ser regulares o de repetición (véase incisos 3.1.47 y 3.1.48 de este proyecto de Norma).
7.4 Los Bancos de Sangre y Centros de Colecta deberán contar con lo siguiente:
7.4.1 El material educativo e informativo referido en el capítulo 6 denominado "Información, consentimientos y atención para donantes y receptores" de este proyecto de Norma para sus donantes;
7.4.2 Procedimientos normalizados de operación para la evaluación de los donantes;
7.4.3 Formatos de historia clínica que cuente con cuestionarios estandarizados en lenguaje comprensible para el público en general, que permitan obtener información relevante acerca de la salud y estilo de vida del candidato a donar sangre o productos sanguíneos.
Estos documentos estarán en todo momento accesibles al personal de salud que participa en el proceso.
7.5 El consultorio donde se efectúe la evaluación médica del donante deberá tener condiciones adecuadas de acceso, iluminación, ventilación, temperatura y asegurar la confidencialidad. Preferentemente deberá ubicarse de manera inmediata o muy cercana a la sala de espera en el caso de establecimientos fijos.
7.6 En caso de colectas externas, los sitios donde se realice la evaluación médica permitirán el aseguramiento de la confidencialidad y deberán contar con el equipo, mobiliario e insumos necesarios.
7.7 La evaluación clínica para obtener sangre o productos sanguíneos de un donante deberá efectuarse de conformidad con lo siguiente:
7.7.1 El médico que la efectúe tendrá capacitación suficiente;
7.7.2 La evaluación la deberá efectuar metódica, cuidadosamente y basada en evidencia clínicas, científicas empleando un lenguaje comprensible y de manera respetuosa para las y los candidatos a donar;
7.7.3 Se llevará a cabo en privado y tendrá carácter confidencial;
7.7.4 Los datos y el resultado de la valoración se registrarán en una historia clínica física y/o electrónica de conformidad con lo señalado en el inciso 20.3.4.1 de este proyecto de Norma, y
7.7.5 Los demás que señala este proyecto de Norma.
7.8 Los Bancos de Sangre y Centros de Colecta deberán analizar los motivos de exclusión temporal y definitiva de los donantes y la prevalencia de los mismos con el fin de detectar las desviaciones en el procedimiento de selección. Estos establecimientos, deberán tener soporte documental de esta actividad.
7.9 La selección de donante y la disposición de la sangre y productos sanguíneos para uso alogénico, deberá efectuarse a través de los procedimientos siguientes:
7.9.1 Identificación del donante;
7.9.2 Evaluación clínica;
7.9.3 Evaluación de resultados de laboratorio;
7.9.4 Autoexclusión del donante, y
7.9.5 Exclusión por terceros.
7.10 Identificación del donante.
El personal asignado del Banco de Sangre o del Centro de Colecta, deberá asegurarse de la identidad de cada donante; para ello, deberá acatar lo siguiente:
7.10.1 Comprobará su identidad con una identificación oficial en original, que esté vigente y que contenga su fotografía;
7.10.2 La comprobación de la identidad se realizará al momento del registro del donante, previamente al inicio de la evaluación clínica e inmediatamente antes de la extracción de la sangre o productos sanguíneos;
7.10.3 Deberá registrar los datos del documento con el que el donante se identifica, para asegurar la trazabilidad del proceso, en caso de que el donante cambie de domicilio deberá notificar al banco de sangre para cualquier notificación de sus estudios, y
7.10.4 Excluirá a quienes no se identifiquen y a aquellos cuyos rasgos fisonómicos no concuerden con los de la fotografía y Clave Única de Registro Poblacional (CURP). En caso de que el donante no porte identificación y su donación sea muy importante, su identidad podrá ser reconocida por el paciente, sus familiares o el médico tratante. En tales casos el responsable del establecimiento o el medico a cargo de la donación lo autorizará por escrito en la historia clínica que al efecto se elabore.
7.11 Evaluación clínica del donante.
7.11.1 Algunos criterios para la selección de los donantes pueden variar de acuerdo al tipo de donación de que se trate, ya sea de sangre total o de algún componente sanguíneo mediante aféresis; para llevar a cabo la selección adecuada de los donantes de sangre se deberán apoyar en los lineamientos establecidos en la Guía Nacional de Criterios para la Selección de Donantes de Sangre y sus Productos sanguíneos para el Uso Terapéutico, en su versión más actual, publicada por el Centro Nacional de Transfusión Sanguínea disponible en https://www.gob.mx/cnts/documentos/recursos-guias-y-documentos-en-medicina-transfusional y en apego al inciso 7.2 de este proyecto de Norma.
7.11.2 Completado el cuestionario de la historia clínica referido en los incisos 7.4.3 y 20.3.4.1 de este proyecto de Norma, deberá ser firmado por el candidato a donar y por el personal médico que lo aplicó, quien verificará que las preguntas relevantes hayan sido adecuadamente contestadas.
7.11.3 La evaluación clínica de los donantes deberá hacerse cada vez que alguien done sangre o productos sanguíneos. La evaluación se efectuará el día de la donación y antes de la extracción.
7.11.4 La exclusión del médico será inapelable.
7.11.5 En caso de controversia el criterio de médico evaluador prevalecerá.
8 Extracción de unidades de sangre y productos sanguíneos para uso alogénico
8.1 Disposiciones comunes.
8.1.1 La sangre para fines transfusionales se obtendrá por extracción venosa. Los productos sanguíneos se podrán obtener por centrifugación o por procesamiento automatizado a partir de unidades de sangre total o por aféresis automatizada con o sin reposición de volumen.
8.1.2 Toda donación deberá registrarse en los libros o sus equivalentes de ingresos y egresos de sangre y productos sanguíneos autorizados y, en su caso, en los sistemas electrónicos (véanse los incisos 20.3.2 al 20.3.2.4 y las tablas 35 y 36 de este proyecto de Norma).
8.1.3 Los Bancos de Sangre y los Centros de Colecta deberán contar con los procedimientos normalizados de operación que se indican a continuación, los cuales deberán estar accesibles al personal de salud que atiende a los donantes:
8.1.3.1 Procedimientos que aseguren la identificación inequívoca de los donantes, los registros, las unidades y las muestras;
8.1.3.2 Procedimientos para efectuar la venopunción, el uso adecuado de los equipos de colecta y la extracción de las unidades y las muestras, y
8.1.3.3 Procedimientos que describan cómo prevenir, tratar y registrar las reacciones o efectos adversos que puedan ocurrir en los donantes.
8.1.4 Los efectos o reacciones adversas relacionadas con la donación deberán mantenerse en un registro y notificarse al Centro Nacional de la Transfusión Sanguínea en el formato o sistema electrónico que para el efecto emita la Secretaría.
8.1.5 Los Bancos de Sangre y los Centros de Colecta deberán registrar cualquier incidente relacionado con el material y los equipos empleados para la colecta de unidades. Asimismo, deberán contar con instrucciones precisas acerca del tipo de incidente y de cómo actuar en cada caso.
8.1.6 A toda donación de sangre o productos sanguíneos deberá de realizarse la búsqueda de agentes transmisibles por transfusión, aunque a la unidad obtenida se le hubiese dado destino final antes de su procesamiento por motivos como el proceso de autoexclusión o cualquier otro evento.
8.1.7 El área física para la extracción y la toma de muestras, tanto de establecimientos fijos como en unidades móviles, deberá tener condiciones adecuadas de acceso, iluminación, ventilación, temperatura, higiene y de seguridad para el donante y el personal de salud.
8.1.8 Los establecimientos fijos y las unidades móviles deberán tener los equipos e insumos necesarios para la extracción de unidades de sangre y, en su caso, para la extracción de productos sanguíneos mediante aféresis; asimismo, deberán contar con los equipos, medicación e insumos necesarios para atender cualquier reacción que ocurriese durante o después de la extracción.
8.1.9 El personal que efectúe la flebotomía deberá usar bata de laboratorio o uniforme, cubre boca o careta de seguridad biológica y guantes, de conformidad a lo que señalen los procedimientos normalizados de operación del establecimiento.
8.1.10 Antes de proceder a la extracción, el personal que hace la flebotomía deberá verificar lo siguiente:
8.1.10.1 Que los datos del donante coincidan con los obtenidos en los documentos utilizados para el proceso de donación;
8.1.10.2 Que los datos de identificación de la bolsa o de los equipos colectores, así como los datos de los tubos para muestras y los registrados en los documentos sean coincidentes;
8.1.10.3 Que las bolsas y equipos colectores se encuentren dentro de su periodo de vigencia, que carezcan de daños, roturas, cambios en su coloración, deterioro o evidencias de contaminación. En caso de cualquier alteración, no deberán ser utilizados. Si la extracción ya se hubiese efectuado, se le dará destino final a la unidad obtenida. De detectarse cualquier defecto en más de una bolsa o equipo colector de un mismo lote, se deberán inmovilizar las bolsas o equipos de ese lote y registrar el incidente, y
8.1.10.4 Las etiquetas con el número de donación que no se hubiesen utilizado deberán ser destruidas conforme a un procedimiento previamente establecido.
8.1.11 Las bolsas colectoras de sangre y los equipos para obtener productos sanguíneos mediante aféresis deberán utilizarse de acuerdo con las instrucciones proporcionadas por el fabricante.
8.1.12 La extracción deberá hacerse empleando métodos asépticos, en sistemas cerrados, evitando la entrada de aire para conservar la esterilidad.
8.1.13 Toda punción para colectar unidades de sangre o productos sanguíneos deberá hacerse conforme a lo siguiente:
8.1.13.1 Se realizará en áreas cutáneas carentes de lesiones;
8.1.13.2 Se utilizarán técnicas asépticas;
8.1.13.3 Se utilizarán técnicas antisépticas validadas, permitiendo que el o los antisépticos empleados ejerzan su acción después de su aplicación. En caso de alergia al yodo, éste podrá sustituirse por clorhexidina.
8.1.14 El anticoagulante y, en su caso, las soluciones aditivas deberán estar en proporción al volumen de sangre o productos sanguíneos que se vaya a extraer.
8.1.15 Para la toma de muestras sanguíneas destinadas a la realización de las determinaciones analíticas y con el fin de asegurase que corresponden a la unidad de sangre o productos sanguíneos colectados, se empleará cualquiera de las metodologías siguientes:
8.1.15.1 Se emplearán bolsas o equipos de colecta que permitan la toma de las muestras antes de que se inicie el proceso de llenado de la unidad de que se trate, o
8.1.15.2 Durante la extracción de las unidades, se llenarán los tubos para las muestras, mediante la bolsa de derivación unida al tubo colector primario y posteriormente continuar con la extracción.
Las muestras se conservarán en tubos con tapón por un lapso mínimo de cuarenta y ocho horas.
8.1.16 El personal que lleva a cabo la extracción deberá registrar en expediente del donante, las actividades realizadas y, en su caso, las reacciones adversas, soluciones empleadas y los medicamentos prescritos.
8.2 Sangre total.
Para la extracción de unidades de sangre total, se deberán observar las disposiciones siguientes:
8.2.1 En toda extracción de sangre total deberán utilizarse bolsas que tengan dos o más bolsas satélites a fin de posibilitar la obtención de productos sanguíneos.
8.2.2 En caso de punción fallida, no deberá intentarse otra punción con el mismo equipo de recolección. Podrá efectuarse un segundo intento siempre y cuando se emplee un equipo nuevo, mientras el donante lo autorice y cuando se prevea que no se excederán los volúmenes a extraer que señala el inciso 8.2.4 de este proyecto de Norma.
8.2.3 Durante el llenado de una unidad de sangre, deberá favorecerse la mezcla con el anticoagulante de la bolsa, la agitación se hará con movimientos de balanceo efectuados aproximadamente cada 45 segundos o preferentemente empleando balanzas con agitación automática que, además, aseguren el volumen sanguíneo neto a recolectar.
8.2.4 El volumen máximo de sangre extraído en cada ocasión deberá ser de 450 ± 10%. No deberá excederse de 10.5 mL por kg de peso corporal del donante o del 13% de su volumen sanguíneo calculado, incluyendo las muestras.
8.2.5 En las bolsas contenedoras más empleadas (bolsas para extracción de sangre), la cantidad de solución anticoagulante y preservadora está calculada para obtener 450 mL ± 10%. Si por razones técnicas no se obtiene un mínimo de 405 mL, se deberá proceder de la manera siguiente:
8.2.5.1 Si el volumen de sangre obtenido es entre 300 y 404 mL, sólo se preparará el concentrado de eritrocitos y se anotará en su etiqueta "unidad de bajo volumen". A los componentes remanentes se le dará destino final, y
8.2.5.2 En el caso de que el volumen de sangre extraído fuese menor de 300 mL, se dará destino final a la unidad.
8.2.6 Para colectar sangre que fuese a destinarse para uso terapéutico de menores de un año de edad, se deben utilizar equipos colectores de sangre con múltiples bolsas satélites que permitan, sin abrir el sistema, repartir el volumen de sangre en cuatro o más bolsas de menor tamaño, o bien, utilizar sistemas de conexión estéril.
8.2.7 Podrán efectuarse variaciones en los volúmenes de recolección, siempre y cuando el procedimiento sea autorizado por escrito por el responsable sanitario del Banco de Sangre o el encargado del Centro de Colecta y la unidad cumpla con los estándares de concentración de hemoglobina por unidad, el hematocrito y leucocitos residuales acorde al tipo de concentrado de eritrocitos obtenido. El documento donde conste la autorización deberá consignar los criterios médicos que justifiquen o sustenten su realización.
8.2.8 Idealmente el tiempo de llenado de una unidad es de entre 8 y 12 minutos. El personal que hace la flebotomía deberá notificar por escrito al área de procesamiento el tiempo de llenado de cada unidad, a fin de que, en caso de extracciones prolongadas, se proceda conforme a lo indicado en los incisos 9.4.1 y 9.6.1.1.1 de este proyecto de Norma.
8.2.9 Al finalizar la extracción, el tubo colector primario de la bolsa deberá sellarse en su extremo distal e inmediatamente el contenido del tubo deberá ser mezclado con la sangre anticoagulada contenida en la bolsa, posteriormente se harán sellos adicionales para preparar segmentos del citado tubo a fin de poder tomar muestras para efectuar las determinaciones analíticas que pudieran requerirse.
8.2.10 En el lapso de un año calendario y bajo criterio del responsable sanitario o del encargado de un centro de colecta el máximo de extracciones sanguíneas practicadas a un donante deberá ser de cuatro si es varón y tres si se trata de una mujer.
8.2.11 Los intervalos mínimos entre extracciones deberán ser los señalados en la Tabla 3 de este proyecto de Norma.
Tabla 3
Intervalos mínimos entre recolecciones de sangre total y otros productos sanguíneos
Procedimientos de extracción |
Intervalo mínimo entre extracciones |
a) Entre dos extracciones de sangre total, o b) Entre una extracción de sangre total y una eritroaféresis de bolsa única (con o sin plasmaféresis o plaquetaféresis). |
Ocho semanas |
c) Entre una donación de sangre total (o eritroféresis de bolsa única) y eritroaféresis de bolsa doble (con o sin plasmaféresis o plaquetaféresis). |
Tres meses |
d) Entre una donación de sangre total y una plaquetaféresis o plasmaféresis sin extracción de eritrocitos. |
Cuatro semanas |
e) Entre dos eritroféresis de bolsa única |
Ocho semanas |
f) Entre dos eritroféresis de bolsa doble |
Seis meses |
g) Entre dos plaquetaféresis sin extracción de eritrocitos, ya sea por colecta sencilla o doble* h) Entre plaquetaféresis (colecta sencilla o doble) y donación de sangre total o eritroféresis de bolsa única con o sin extracción de multicomponentes |
Dos semanas |
i) Entre donación de sangre total o eritroféresis de bolsa única y plaquetaféresis de colecta sencilla o doble |
Cuatro semanas |
j) Entre dos plasmaféresis sin necesidad de reposición de volumen a k) Entre dos granulocitaféresis |
Dos semanas |
l) Entre una donación de células progenitoras hematopoyéticas de sangre periférica y una donación de sangre total m) Entre una donación de donación de células progenitoras hematopoyéticas de sangre periférica y, plaquetaféresis, eritroféresis, plasmaféresis, linfocitos o granulocitos |
Seis meses |
n) Entre una donación de células progenitoras hematopoyéticas por médula ósea y sangre total** o) Entre una donación células progenitoras hematopoyéticas por médula ósea y una plaquetaféresis, eritroféresis, plasmaféresis, linfocitos o granulocitos** |
Un año |
*Bajo criterio médico, en casos especiales como refractariedad plaquetaria, sensibilización en contra de antígenos plaquetarios o leucocitarios específicos o necesidad de transfusión de preparados con plaquetas HLA compatibles, el intervalo entre donación de dos plaquetaféresis podrá disminuir a 48 horas, siempre y cuando no se exceda de dos procedimientos por semana. a Bajo criterio médico, se podrá disminuir el intervalo a 48 horas como mínimo, siempre y cuando no sobrepase más de un litro de volumen de extracción a la semana.** En caso de que el paciente trasplantado requiere algún componente sanguíneo del donante de células progenitoras hematopoyéticas, antes de que se cumplan los periodos señalados en los incisos l) a o), el tiempo de donación del mismo, quedará a criterio del médico responsable sanitario del Banco de Sangre y del médico tratante del paciente receptor.
8.3 Extracción de productos sanguíneos mediante métodos aféresis.
Para la extracción de cualquier componente sanguíneo mediante métodos de aféresis, se deberán observar las disposiciones siguientes:
a) Antes de cada extracción se deberá comprobar que el donante cumpla con los parámetros de laboratorio mínimos, de acuerdo con el componente sanguíneo que vaya a colectarse, de conformidad a lo señalado en la tabla 4 de este proyecto de Norma;
b) En todo momento del procedimiento el volumen sanguíneo extracorpóreo no deberá exceder del 13% del volumen sanguíneo total.
8.3.1 Eritroaféresis.
Mediante este procedimiento se podrá obtener el equivalente a una o dos unidades de concentrado de eritrocitos.
Para estos procedimientos se deberá acatar lo siguiente:
a) La cantidad de glóbulos rojos extraídos no excederá del volumen teórico que haga que el nivel de hemoglobina del donante caiga por debajo de 110 g/L, y
b) La pérdida anual de eritrocitos no deberá exceder el equivalente a la masa eritrocítica contenida en cuatro unidades de sangre para donantes masculinos y tres unidades de sangre para donantes femeninos.
8.3.1.1 Eritroaféresis, bolsa única.
Los intervalos mínimos entre extracciones de eritroaféresis de bolsa única deberán ser los señalados en la Tabla 4 de este proyecto de Norma.
Tabla 4
Intervalos mínimos entre recolecciones de eritrocitos mediante aféresis
Procedimientos de extracción |
Intervalo mínimo entre extracciones |
a) Entre dos eritroaféresis de bolsa única. |
Ocho semanas |
b) Entre una eritroaféresis de bolsa única o falla en regresar al donante sus glóbulos rojos durante una aféresis y el siguiente procedimiento de aféresis que no incluya eritroaféresis. |
Cuatro semanas |
8.3.1.2 Eritroaféresis, bolsa doble.
Cuando se pretenda obtener dos unidades de concentrado de eritrocitos, se deberá observar lo siguiente:
8.3.1.2.1 El volumen sanguíneo del donante deberá ser mayor de 5 L. Generalmente este requisito se cumple cuando el donante pesa 70 Kg o más en ausencia de obesidad
8.3.1.2.2 Los intervalos mínimos entre donaciones mediante eritroaféresis de bolsa doble u otras extracciones, deberán ser los señalados en la Tabla 5 de este proyecto de Norma.
Tabla 5
Intervalos mínimos entre recolecciones de eritrocitos mediante aféresis bolsa doble y otras recolecciones
Procedimientos de extracción |
Intervalo mínimo entre extracciones |
a) Entre la donación de una unidad de sangre total o una unidad de eritroaféresis y una eritroaférisis de doble bolsa. |
Tres meses |
b) Entre una extracción doble de concentrado de eritrocitos y una donación de sangre total o una eritroaféresis de bolsa única o doble. |
Seis meses |
8.3.2 Plaquetaféresis.
8.3.2.1 Mediante plaquetaféresis podrán obtenerse una o más cosechas de concentrados de plaquetas. El equipo automatizado para la colecta de plaquetas deberá predeterminarse a fin de evitar que la cuenta de plaquetas del donante descienda por abajo de 100 x 109/L.
8.3.2.2 Los intervalos mínimos entre donaciones serán los que indica la Tabla 6 de este proyecto de Norma.
Tabla 6
Intervalos mínimos entre colectas de plaquetas mediante aféresis de bolsa sencilla o doble y otras colectas
Procedimientos de extracción |
Intervalo mínimo entre extracciones |
a) Entre dos donaciones de plaquetas óptimamente efectuadas de colecta sencilla o doble. |
Dos semanas (véase nota) |
b) Entre una donación de plaquetas óptimamente efectuada (colecta sencilla o doble) y una donación de sangre total o una eritroaféresis de bolsa única, combinada o no con recolección de plasma y plaquetas. |
Dos semanas |
c) Entre una donación de sangre total o una eritroaféresis de colecta sencilla y una plaquetaféresis de bolsa sencilla o doble. |
4 semanas |
Nota: Bajo criterio médico, en casos especiales, tales como requerimientos transfusionales en receptores sensibilizados a antígenos plaquetarios o leucocitarios específicos, el intervalo referido en el inciso a) de esta tabla podrá reducirse a 48 horas, siempre y cuando no se hagan más de dos plaquetaféresis óptimamente efectuadas en el lapso de una semana.
8.3.2.3 Salvo en circunstancias excepcionales, cuando por plaquetaféresis sucesivas las pérdidas acumuladas de eritrocitos superen los 200 mL, o se extraiga una unidad de concentrado de eritrocitos adicional, o resulte imposible retornar al donante sus glóbulos rojos, deberá dejarse transcurrir un plazo de al menos 4 semanas antes de realizar otra plaquetaféresis, siempre y cuando en el nuevo proceso no se extraigan eritrocitos, en cuyo caso el intervalo será de 8 semanas.
8.3.2.4 El máximo de plaquetaféresis de colecta sencilla o doble no deberá exceder de 24 procedimientos en el lapso de un año.
8.3.3 Plasmaféresis.
Para efectuar una plasmaféresis se deberá observar lo siguiente:
8.3.3.1 El volumen plasmático máximo extraído por sesión no deberá exceder de 600 mL, excluyendo el volumen del anticoagulante, o del 16% del volumen sanguíneo total, en ausencia de reposición volumétrica.
8.3.3.2 La frecuencia máxima será de un procedimiento cada dos semanas. En circunstancias excepcionales, y bajo criterio médico, este tiempo podrá acortarse a un intervalo mínimo de 48 horas, siempre y cuando no se sobrepase de un volumen de 1 litro en el lapso de una semana.
8.3.3.3 El lapso mínimo entre una plasmaféresis y una donación de sangre total convencional o una eritroaféresis de bolsa única (combinada o no con recolección de plasma y plaquetas) será de 48 horas.
8.3.3.4 En caso de que a un donante de plasmaféresis no sea posible retornarle sus eritrocitos o se extraiga una unidad de glóbulos rojos adicional, se diferirá la siguiente plasmaféresis por 8 semanas.
La extracción plasmática máxima en el lapso de un año no deberá exceder de 15 litros.
8.3.4 Granulocitaféresis.
Para efectuar este procedimiento se deberá acatar lo siguiente:
8.3.4.1 En cada extracción no deberá excederse de un volumen de 500 mL.
8.3.4.2 Entre cada procedimiento óptimamente realizado deberá haber un lapso mínimo de dos semanas.
8.3.4.3 En caso de realizar más de una granulocitaféresis en el lapso de cuatro semanas y de emplearse agentes que favorezcan la sedimentación, éstos se emplearán en dosis sucesivamente decrecientes, a fin de evitar toxicidad.
8.3.4.4 En un donante no deberá excederse de doce procedimientos óptimamente realizados en el lapso de un año.
8.3.4.5 No se administrarán esteroides a aquellos donantes con enfermedades crónicas que puedan exacerbarse o que tengan cualquier contraindicación para su utilización, tales como hipertensión arterial, úlcera péptica o diabetes mellitus.
8.3.5 Aféresis de multicomponentes.
Para la realización de una aféresis de multicomponentes se deberá observar lo siguiente:
8.3.5.1 Deberán cumplirse los requisitos de cada tipo de donación en lo referente a valores de hematimetría previos a la extracción (véase Tabla 9 de este proyecto de Norma) y el intervalo entre donaciones.
8.3.5.2 El volumen total extraído de glóbulos rojos, plasma y plaquetas no deberá ser mayor de 600 mL (excluido el anticoagulante) o del 13% del volumen sanguíneo del donante. De exceder estos límites deberá hacerse remplazo volumétrico con soluciones adecuadas.
8.3.5.3 La cantidad de glóbulos rojos extraída no excederá del volumen teórico que haga que el nivel de hemoglobina del donante caiga por debajo de 110 g/L.
8.3.5.4 El periodo de tiempo de espera entre una donación de plaquetaféresis, eritroféresis, plasmaféresis, linfocitos, granulocitos o de sangre total y una donación de células progenitoras hematopoyéticas en sangre periférica, será de seis meses.
8.3.5.5 El periodo de tiempo de espera entre una donación de plaquetaféresis, eritroféresis, plasmaféresis, linfocitos, granulocitos o de sangre total y una donación de células progenitoras hematopoyéticas por médula ósea, será de doce meses.
8.3.5.6 El periodo de tiempo de espera entre una donación de células progenitoras en sangre periférica y de plaquetaféresis, eritroféresis, plasmaféresis, linfocitos, granulocitos o de sangre total y una donación de células progenitoras hematopoyéticas en sangre periférica, será de seis meses.
8.3.5.7 El periodo de tiempo de espera entre una donación de células progenitoras hematopoyéticas por médula ósea y una donación de plaquetaféresis, eritroféresis, plasmaféresis, linfocitos, granulocitos o de sangre total, será de doce meses.
8.3.5.8 En caso de que el paciente trasplantado requiera algún componente sanguíneo del donante de células progenitoras hematopoyéticas, antes de que se cumplan los periodos señalados en los puntos 7.3.5.4-7.3.5.7; el tiempo de donación del mismo, quedará a criterio del médico responsable sanitario del Banco de Sangre y del médico tratante del paciente receptor.
9 Procesamiento, conservación, vigencia y control de calidad de las unidades de sangre y productos sanguíneos
9.1 Disposiciones comunes:
9.1.1 Los Bancos de Sangre, los Centros de Colecta, Centros de Procesamiento de Sangre, Centros de Distribución de Sangre y Productos sanguíneos, Centros de Calificación Biológica y Servicios de Transfusión Hospitalaria, en el ámbito de las funciones que les autoriza la Secretaría de Salud, deberán observar lo siguiente:
9.1.1.1 Deberán tener procedimientos normalizados de operación relativos al procesamiento, condiciones adecuadas de almacenamiento y temperatura de conservación de la sangre, productos sanguíneos, reactivos y muestras, que incluyan las instrucciones a seguir en caso de falla equipos, instrumentos, materiales, suministro eléctrico o cualquier otra eventualidad, y
9.1.1.2 Registrar cualquier tipo de incidente relacionado con las actividades a que se refiere el inciso anterior.
9.1.2 Dentro de los compartimentos de los equipos para la conservación de unidades de sangre, productos sanguíneos, mezclas de componente sanguíneos, reactivos y muestras, deberá haber una distribución, separación y señalización suficiente, conforme a lo que se indica a continuación:
9.1.2.1 Unidades no procesadas o aún no estudiadas;
9.1.2.2 Unidades o mezclas procesadas y estudiadas;
9.1.2.3 Unidades o mezclas seleccionadas para determinados pacientes;
9.1.2.4 Unidades o mezclas destinadas para uso autólogo;
9.1.2.5 Unidades o mezclas para destino final;
9.1.2.6 Muestras sanguíneas, y
9.1.2.7 En su caso, reactivos.
Los compartimentos de refrigeradores, congeladores, ultracongeladores o cámaras frías destinados a la conservación de unidades de sangre, productos sanguíneos, mezclas de productos sanguíneos, reactivos y muestras, no se emplearán para la conservación de alimentos, bebidas o materiales contaminantes.
9.1.3 Las unidades de sangre y productos sanguíneos aún no estudiadas o con alguna falla técnica o alteración en las determinaciones analíticas deberán mantenerse bajo estricta custodia, separadas del resto de las existencias y a las temperaturas de conservación adecuadas hasta que se hubieran concluido o, en su caso, enmendado los estudios pertinentes y haber obtenido resultados confiables en las determinaciones analíticas. Sólo bajo estas condiciones podrá autorizarse su uso terapéutico o su destino final.
9.1.4 Durante el procesamiento de las unidades de sangre total y productos sanguíneos, o al hacer mezclas de éstas, se deberá mantener su esterilidad, para lo cual se emplearán métodos cerrados, soluciones estériles y libres de pirógenos y, en su caso, conectores estériles (véase el inciso 9.1.6 de este proyecto de Norma).
9.1.5 El plasma, la solución de cloruro de sodio al 0.9%, las soluciones aditivas y la albúmina humana son los únicos que pueden emplearse para reconstituir o, en su caso, resuspender unidades de productos sanguíneos.
9.1.6 Es preferible el empleo de equipos que permitan la transferencia de los productos sanguíneos y de las soluciones a otros contenedores al tiempo que conservan el sistema cerrado o empleando sistemas de conexión estéril, puesto que bajo estas circunstancias no se altera la vigencia de las unidades.
9.1.7 Además de los controles de calidad para las unidades de sangre y productos sanguíneos que se especifican en este capítulo, las unidades deberán someterse a las determinaciones analíticas que se señalan en el capítulo 10 denominado "Determinaciones analíticas" de este proyecto de Norma.
9.1.8 Los establecimientos que procesen sangre total y sus componentes (Bancos de Sangre y Centros de Procesamiento de Sangre) deberán disponer de métodos para limitar y detectar la contaminación para cada componente sanguíneo, estableciendo un límite crítico esperable de contaminación. En caso de observarse cualquier desviación se deberá aumentar el tamaño de la muestra y revisar los procedimientos de extracción y procesamiento.
9.1.9 Se deberá dar destino final a las unidades de sangre, productos sanguíneos o mezcla de componentes en los casos siguientes:
9.1.9.1 Cuando hubieran llegado a su límite de vigencia;
9.1.9.2 Cuando su sistema haya sido abierto en condiciones inciertas de esterilidad, y
9.1.9.3 Los demás que señala este proyecto de Norma.
9.2 Sangre, concentrados de eritrocitos y sangre reconstituida.
9.2.1 El intervalo entre la extracción y el procesamiento de la sangre total se llevará a cabo a la brevedad posible con el fin de conservar los efectos terapéuticos de cada uno de los componentes que la (véase incisos 9.4.3.1, 9.4.3.2, 9.4.3.3 y 9.6.1.1.2 de este proyecto de Norma), durante este intervalo las unidades se mantendrán a temperaturas por debajo de los +24º C, entre +2° y +6° C o bien, entre +2° y +10° C, cuando vayan a transportarse. En cualquier caso, el tiempo máximo de procesamiento no excederá de 24 horas.
9.2.2 Durante su almacenamiento las unidades de sangre total y concentrados de eritrocitos deberán conservarse entre +2º C y +6º C; en estas condiciones su periodo de vigencia varía de acuerdo con el método de procesamiento y a la solución que contienen, conforme a señalado en la Tabla 7 de este proyecto de Norma.
Tabla 7
Vigencia de las unidades de sangre total y de los concentrados de eritrocitos
Unidad |
Anticoagulante o solución que contienen |
Vigencia máxima |
Temperatura de conservación |
En sistemas cerrados |
|||
Sangre y concentrados de eritrocitos |
CPDA |
35 días a partir de la extracción |
+2º C y + 6º C |
CPD con solución aditiva |
42 días a partir de la extracción |
||
En sistemas abiertos |
|||
Sangre y concentrados de eritrocitos |
CPDA (hay tipo 1 tipo 2) y CPD con solución aditiva |
24 horas a partir de la apertura del sistema |
+2º C y + 6º C |
Seis horas a partir de la apertura del sistema |
+6º y +10º C |
||
9.2.3 Los requisitos de calidad que deberán tener las unidades de sangre total, que fuesen a emplearse con fines transfusionales, verificadas con el número de unidades y la frecuencia indicados, se muestran en la Tabla 8 de este proyecto de Norma.
Tabla 8
Requisitos de calidad que deberán reunir el 100% de las unidades de sangre total probadas
Parámetro |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
450 mL ± 10%, excluyendo el anticoagulante |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemoglobina (sólo en las unidades que fuesen a usarse en transfusión) |
≥45 g por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemoglobina en caso de utilizarse filtros de última generación |
≥ 43 g por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Leucocitos residuales en caso de utilizarse filtros de última generación |
< 1 x106 leucocitos / unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemólisis al término de la vigencia (véase Nota al pie de esta tabla) |
< 0.8% de la masa eritrocítica |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Control bacteriológico, únicamente cuando las unidades de sangre total fuesen a transfundirse sin procesar. |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Nota: La hemólisis al término de la vigencia podrá evaluarse mediante técnicas validadas tales como cuantificación de hemoglobina libre o por determinación de azidametahemoglobina.
*CEP= Control Estadístico de Proceso.
9.3 Concentrados de eritrocitos.
9.3.1 Los concentrados de eritrocitos se utilizan para suplir pérdidas sanguíneas y para tratamiento paliativo de algunas anemias.
9.3.2 Preferentemente las unidades de concentrados de eritrocitos deberán prepararse removiendo la capa leucoplaquetaria.
9.3.3 Los requisitos de calidad que deberán tener los concentrados de eritrocitos, verificados con el número de unidades y la frecuencia indicada, se muestran en la Tabla 9 de este proyecto de Norma.
Tabla 9
Requisitos de calidad que deben reunir el 100% de las unidades de concentrados de eritrocitos probadas
9.3.3.1 Concentrado de eritrocitos |
||
Parámetro |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
280 ± 50 mL |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hematocrito (véase nota al pie de esta tabla) |
65 - 75% |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemoglobina |
>45 g/U |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
< 0.8% de la masa eritrocítica |
|
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
9.3.3.2 Concentrado de eritrocitos sin capa leucoplaquetaria |
||
Parámetro |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
250 ± 50 mL |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hematocrito (véase nota al pie de esta tabla) |
65 - 75% |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemoglobina |
>43 g por unidad |
|
Leucocitos |
<1.2 x 106por unidad (en el 95% de las unidades probadas) |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
<0.8% de la masa eritrocítica |
|
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
9.3.3.3 Concentrado de eritrocitos en solución aditiva |
||
Parámetro |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
(Depende del proceso obtenido por CEP*) |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hematocrito (véase nota al pie de esta tabla) |
50 - 70% |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemoglobina |
> 45 g por unidad |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
< 0.8% de la masa eritrocítica |
|
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
9.3.3.4 Concentrado de eritrocitos sin capa leucoplaquetaria en solución aditiva |
||
Parámetro |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
(Depende del proceso obtenido por CEP*) |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hematocrito (véase nota al pie de esta tabla) |
50 - 70% |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemoglobina |
>43 g por unidad |
|
Leucocitos residuales |
<1.2 x 109 por unidad (en el 90% de las unidades probadas) |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
< 0.8% de la masa eritrocítica |
|
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
9.3.3.5 Concentrado de eritrocitos leucodepletados en solución aditiva |
||
Parámetro |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
(Depende del proceso y sistema utilizado, debe ser obtenido por CEP*) |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hematocrito (véase nota al pie de esta tabla) |
50 - 70% |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hemoglobina |
>40 g por unidad |
|
Leucocitos residuales |
<1.0 x 106 por unidad (en el 90% de las unidades probadas) |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
<0.8% de la masa eritrocítica |
|
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Nota: La viabilidad de los eritrocitos puede afectarse cuando el hematocrito de la unidad excede al 80%.
*CEP= Control Estadístico de Proceso.
9.3.4 Concentrados de eritrocitos lavados. En su preparación y conservación se observará lo siguiente:
9.3.4.1 Se prepararán a partir de concentrados de eritrocitos a los que se ha efectuado una máxima remoción del plasma o solución aditiva y de la capa leucoplaquetaria;
9.3.4.2 Se harán lavados sucesivos con solución salina isotónica al 0.9% que tenga una temperatura entre +2º C y +6º C y la centrifugación se hará en centrífugas con temperatura controlable;
9.3.4.3 Al finalizar el lavado los eritrocitos se suspenderán en solución salina isotónica al 0.9% o una mezcla de solución salina y solución aditiva, y
9.3.4.4 La temperatura de conservación deberá ser entre +2º C y +6º C hasta su uso terapéutico.
9.3.5 Los requisitos de calidad que deberán tener los concentrados de eritrocitos lavados, verificados en las unidades procesadas, se muestran en la Tabla 10 de este proyecto de Norma.
Tabla 10
Requisitos de calidad que deben reunir el 100% de las unidades de concentrados de eritrocitos lavados con solución salina isotónica al 0.9%
Parámetro a verificar |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
Variable según el método empleado |
Todas las unidades |
Observación del sobrenadante |
Incoloro después del último lavado |
|
Hematocrito final |
(40%-70%) |
|
Hemoglobina |
>40 g por unidad |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
<0.8% de la masa eritrocítica |
|
Proteínas en el sobrenadante final (véase Nota al pie de esta tabla) |
<0.5 g por unidad |
|
Control bacteriológico al final del procesamiento. |
Sin desarrollo |
Nota: La cantidad indicada de proteínas en el sobrenadante asegura que el contenido de inmunoglobulina tipo A sea menor de 0.2 mg por unidad.
9.3.5.1 La transfusión de los eritrocitos lavados deberá ser tan pronto como sea posible después de su preparación, sin que el intervalo exceda de 24 horas y que se conserven entre +2º C y +6º C.
9.3.6 Concentrados de eritrocitos congelados.
9.3.6.1 Las unidades de concentrados de eritrocitos que se pretendan congelar no deberán exceder de los siete días que siguen a su extracción.
9.3.6.2 A los concentrados de eritrocitos se les deberá agregar glicerol al 40% o al 20% como crioprotector y mantenerse constantemente a las temperaturas que se indican a continuación:
9.3.6.2.1 Con alta concentración de glicerol, en congeladores eléctricos, entre -70º C y -80º C, o
9.3.6.2.2 Con baja concentración de glicerol, en vapor de nitrógeno líquido, entre -140º C y -150º C.
9.3.6.3 Los concentrados de eritrocitos en congelación tendrán una vigencia máxima de 10 años, siempre y cuando se garantice el mantenimiento constante de las temperaturas referidas en el inciso que antecede.
9.3.6.4 Dado el tiempo prolongado de conservación se deberán conservar muestras de suero, plasma o ambos obtenidas durante la recolección de la unidad para los estudios posteriores que fuesen necesarios en caso de nuevos marcadores de enfermedades transmisibles.
9.3.6.5 Para utilizar los concentrados de eritrocitos congelados deberán descongelarse y lavarse mediante recambios con solución salina isotónica al 0.9% y suspenderse en la citada solución o en solución aditiva para eritrocitos.
9.3.6.6 Los requisitos de calidad que deberán tener los concentrados de eritrocitos descongelados, verificados con el número de unidades y la frecuencia que se indican, se muestran en la Tabla 11 de este proyecto de Norma.
Tabla 11
Requisitos de calidad que deben reunir el 100% de las unidades de concentrados de eritrocitos descongelados y reconstituidos o resuspendidos probadas
Parámetro a verificar |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
Según el método |
Todas las unidades procesadas |
Observación del sobrenadante |
Incoloro después del último lavado |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
<0.2 g por unidad |
|
Hematocrito |
65% - 75% |
|
Hemoglobina |
>36 g por unidad |
|
Osmolaridad de la solución final |
<340 mOsm/L |
Todas las unidades procesadas. |
Control bacteriológico al final del procesamiento. |
Sin desarrollo |
Todas las unidades procesadas |
9.3.6.7 Los concentrados de eritrocitos descongelados, lavados y resuspendidos deberán conservarse entre +2º C y +6º C y transfundirse en cuanto sea posible, sin exceder de 24 horas cuando el procesamiento se ha hecho en sistema abierto.
9.3.7 Concentrado de eritrocitos obtenido por aféresis.
9.3.7.1 En una sesión de aféresis se podrán colectar uno o dos concentrados de eritrocitos. Durante o después del procedimiento se podrá añadir una solución aditiva, en el volumen recomendado por el fabricante del equipo de aféresis.
9.3.7.2 Las condiciones de almacenamiento y caducidad de los concentrados de eritrocitos obtenidos por aféresis de las soluciones aditivas y los demás métodos de procesamiento que se usen (véase la tabla 13 de este proyecto de Norma).
9.3.7.3 Los requisitos de calidad que deberán tener los concentrados de eritrocitos obtenidos por aféresis, verificados con el número de unidades y la frecuencia que se indican, se muestran en la Tabla 12 de este proyecto de Norma.
Tabla 12
Requisitos de calidad que deben reunir el 100% de las unidades de concentrados de eritrocitos obtenidos por aféresis probadas
Parámetro a verificar |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
Según el sistema usado |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hematocrito |
65% - 75% |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Hematocrito, en caso de agregar solución aditiva |
50% - 70% |
|
Hemólisis al término de la vigencia (véase Nota al pie de la tabla 14) |
< 0.8% de la masa eritrocítica |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
9.3.8 Concentrados de eritrocitos rejuvenecidos.
Para los procedimientos de rejuvenecimiento de los concentrados de eritrocitos, se deberá observar lo siguiente:
9.3.8.1 Se emplearán métodos con capacidad conocida de restaurar a niveles normales o más altos del 2,3- difosfoglicerato y el trifosfato de adenosina;
9.3.8.2 Se seguirán las instrucciones escritas proporcionadas por el fabricante de la solución rejuvenecedora;
9.3.8.3 Los concentrados de eritrocitos elegibles para el procedimiento estarán conservados entre +2º y +6º C y no se emplearán aquéllos que sobrepasen de los tres días que anteceden a su límite de vigencia, y
9.3.8.4 Previo a su suministro para uso terapéutico deberán lavarse mediante recambios sucesivos con solución salina isotónica al 0.9% y transfundirse en un lapso que no exceda de 24 horas después del lavado, o bien, glicerolarse y congelarse.
9.3.9 Sangre reconstituida.
La sangre reconstituida deberá reunir los requisitos siguientes:
9.3.9.1 Se preparará bajo condiciones de esterilidad en campana de flujo laminar o con el uso de conectores estériles;
9.3.9.2 Dependiendo de su indicación, el plasma y el concentrado de eritrocitos empleados en su elaboración podrán ser del mismo donante o de donantes distintos;
9.3.9.3 El concentrado de eritrocitos y el plasma podrán no ser coincidentes en el grupo sanguíneo AB0 y Rh (D) siempre y cuando exista compatibilidad entre ellos, de manera que la mezcla no provoque hemólisis de los eritrocitos;
9.3.9.4 El hematocrito final después de efectuada la reconstitución será de entre 40 y 50%, salvo en indicaciones especiales tales como exsanguineotransfusión y transfusión intrauterina (véanse incisos 12.8.3 y 12.9.1.1 de este proyecto de Norma, respectivamente);
9.3.9.5 El volumen final dependerá directamente del volumen del concentrado de eritrocitos y del volumen del plasma utilizado para hacer la reconstitución;
9.3.9.6 El plasma empleado para la reconstitución deberá carecer de anticuerpos irregulares, y
9.3.9.7 La unidad de eritrocitos empleada en la reconstitución deberá carecer de anticuerpos irregulares a menos que se empleen eritrocitos en solución aditiva o lavados.
9.4 Plaquetas.
9.4.1 Los concentrados de plaquetas se podrán obtener por procesamiento o método automatizado a partir de sangre fresca a partir del plasma rico en plaquetas o de la capa leucoplaquetaria (proceso de doble centrifugación), o bien, mediante aféresis automatizada. Solo se obtendrán plaquetas de unidades de sangre total cuando el tiempo de extracción de la sangre del donante no hubiese excedido de 12 minutos.
9.4.2 Los preparados con plaquetas se mantendrán preferentemente dentro de equipos de almacenaje que tengan las características siguientes:
9.4.2.1 Armario incubador cerrado con control de temperatura entre +20º C y +24º C, y
9.4.2.2 Que en su interior haya superficies que realicen movimientos horizontales suaves, oscilatorios, de no más de 70 revoluciones por minuto (rpm), que mantengan las plaquetas en agitación constante para mantener un mezclado del contenido de la bolsa y sin exceder la capacidad de unidades a almacenar recomendada por el fabricante del armario, con el fin de permitir un intercambio gaseoso a través de la pared de la bolsa y evitar que éstas se plieguen.
De no contarse con armario incubador, los preparados con plaquetas se mantendrán en agitadores abiertos, con movimientos oscilatorios o giratorios, en áreas capaces de mantener la temperatura requerida de manera constante y verificable.
9.4.3 Para optimizar la viabilidad y funcionalidad de las plaquetas en unidades o mezclas, se deberá observar lo siguiente:
9.4.3.1 Cuando se obtienen por procesamiento de unidades de sangre total:
9.4.3.2 La sangre tendrá menos de 24 horas de haberse extraído, siempre y cuando se hubiese conservado entre +20º C a +24º C, y
9.4.3.3 Las unidades de sangre deberán centrifugarse dentro del rango temperatura de +20º C a +24° C;
9.4.3.4 Las bolsas contenedoras de plaquetas deberán ser suficientemente permeables a gases, a fin de garantizar la disponibilidad de oxígeno. Para los mismos fines las bolsas se colocarán a manera que haya circulación de aire entre las mismas, y
9.4.3.5 Son condiciones adecuadas para la conservación de plaquetas cuando:
9.4.3.6 Se mantienen a una temperatura entre +20º C a +24º C, preferiblemente en agitación suave continua;
9.4.3.7 La concentración de plaquetas es menor a 1.5 x 109 por mililitro de plasma o mezcla de plasma-solución nutritiva, y
9.4.3.8 El pH de la preparación se mantiene constantemente entre 6.4 y 7.4.
9.4.4 Para el procesamiento y conservación de los preparados de plaquetas lavadas, se observará lo siguiente:
9.4.4.1 Se prepararán a partir de concentrados o mezclas de plaquetas a los que se ha efectuado una máxima remoción del plasma;
9.4.4.2 Se harán lavados sucesivos con solución salina isotónica al 0.9 % o soluciones especiales para tal fin, a una temperatura entre +20º C y +24º C y en centrífugas con temperatura controlable, y
9.4.4.3 Al finalizar el lavado se suspenderán en solución salina isotónica al 0.9% o solución salina con amortiguador y se mantendrán entre +20º C y +24º C hasta su utilización terapéutica.
9.4.5 La vigencia asignada a las unidades o mezclas de plaquetas deberá ser la que señala la Tabla 13 de este proyecto de Norma, dependiendo de las variables que se señalan.
Tabla 13
Vigencia de unidades de plaquetas recuperadas de la sangre total, mezclas de unidades y unidades obtenidas por aféresis
Variables |
Vigencia |
|
Otras condiciones |
Temperatura de conservación |
|
a) En sistema cerrado, en agitación continua suave; |
+20º C a +24º C |
Cinco días después de la donación, ampliable hasta 7 días si se emplean sistemas de reducción bacteriana o métodos de detección de contaminación bacteriana. |
b) En sistema cerrado, sin agitación; |
+20º C a +24º C |
Máximo 24 horas después de la donación. |
c) Plaquetas lavadas (descongeladas o no), en agitación continua suave; |
+20º C a +24º C |
Máximo seis horas a partir de la apertura del sistema o del procedimiento (es preferible transfundirlas en el menor tiempo posible). |
d) Plaquetas con escaso volumen de plasma, y |
||
e) Plaquetas en sistemas abiertos. |
||
Nota: Los procedimientos de irradiación o filtrado para leucodepleción no altera la vigencia asignada a los preparados de plaquetas.
9.4.6 Concentrados de plaquetas unitarios y mezclas de unidades.
9.4.6.1 Los establecimientos que preparen plaquetas deberán verificar los requisitos de calidad de los concentrados y mezclas de plaquetas con la cantidad de productos y con la frecuencia que señala la Tabla 14 de este proyecto de Norma.
Tabla 14
Requisitos de calidad que deben reunir el 100% de las unidades o mezclas de plaquetas recuperadas del plasma rico en plaquetas o de la capa leucoplaquetaria probadas
Parámetro a verificar |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Inspección de la unidad |
Ausencia de agregados plaquetarios |
Todas las unidades preparadas en el mes |
Volumen por unidad |
El validado para contener 0.6 x 1011 plaquetas |
|
Contenido de plaquetas |
>6.0 x 1010 por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Leucocitos residuales en unidades recuperadas de plasma rico en plaquetas |
<0.2 x 109 por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Leucocitos residuales en unidades recuperadas de la capa leucoplaquetaria |
<0.05 x 109 por unidad |
|
Leucocitos residuales, en unidades o mezclas de plaquetas tras leucodepleción por filtración |
<0.2 x 106 por unidad (en el 90 % de las unidades probadas) |
|
pH al término de su vigencia |
>6.4 |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
9.4.6.2 A cualquier preparado de plaquetas (unidades recuperadas, mezclas o unidades obtenidas por aféresis) que hubiese llegado a su límite de vigencia o que muestre agregados plaquetarios se le deberá dar destino final.
9.4.7 Concentrados de plaquetas obtenidos por aféresis.
9.4.7.1 Las unidades de plaquetas obtenidas por aféresis pueden reducir los riesgos de aloinmunización a antígenos leucocitarios humanos y de transmisión viral al disminuir el número de exposiciones alogénicas; así mismo, constituyen un tratamiento efectivo para pacientes previamente aloinmunizados.
9.4.7.2 Las unidades de plaquetas obtenidas por aféresis pueden contener dosis terapéuticas efectivas en adultos utilizando mezcla de plasma (30 a 40%) y una solución aditiva (60 a 70%).
9.4.7.2.1 Conteniendo un mínimo de 2 x 1011 plaquetas.
9.4.7.2.2 Conteniendo menos de 1 x 106 leucocitos.
9.4.7.3 Las unidades de plaquetas obtenidas por aféresis podrán antes de su conservación recibir un tratamiento de inactivación de patógenos validado, con la finalidad de reducir el riesgo de transmisión de virus envueltos (VIH, VHB, VHC), algunas bacterias (excepto esporas bacterianas) dependiendo de la tecnología utilizada.
Dependiendo del procedimiento, algunos tratamientos de inactivación de patógenos han mostrado que puede inactivar linfocitos, por lo que, la irradiación puede no ser necesaria para prevenir la enfermedad injerta contra hospedero.
9.4.7.3.1 Conteniendo un mínimo de 2 x 1011 plaquetas.
Conteniendo menos de 1 x 106 leucocitos.
9.4.7.4 Se deberá verificar los requisitos de calidad de las unidades de plaquetas obtenidas por aféresis, con la cantidad de unidades y la frecuencia que señala la Tabla 15 de este proyecto de Norma.
Tabla 15
Requisitos de calidad que deben reunir el 100% de las unidades de plaquetas obtenidas por aféresis probadas
Parámetro a verificar |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Inspección de la unidad |
Ausencia de agregados plaquetarios |
Todas las unidades obtenidas en el mes |
Volumen (depende del contenido de plaquetas) |
Calcular de acuerdo al contenido de plaquetas y el sistema utilizado. |
|
Contenido de plaquetas |
Unidad estándar ≥ 200 x 109 Para uso en neonatos o infantes ≥ 0.5 x 1011 (en el 90% de las unidades) |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Leucocitos residuales en unidades leucorreducidas (véase Nota al pie de esta tabla) |
p < 1.0 x 10 9 por unidad (en el 90% de las unidades) |
|
Leucocitos residuales en unidades leucodepletadas por uso de filtros de última generación |
< 1.0 x 106 por unidad |
|
pH al término de su vigencia |
>6.4 |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
HLA o HPA (en caso de requerirse) |
Tipificación |
Cada vez que se requiera. |
Control bacteriológico |
Sin desarrollo |
Establecer frecuencia de acuerdo a guía de control de calidad microbiológico de componentes plaquetarios ** |
Nota: Con algunos equipos de aféresis, el contenido de leucocitos residuales puede ser mucho más bajo.
** Guía de control de calidad microbiológico de componentes plaquetarios para incrementar la seguridad en su uso clínico en México (septiembre 2020) CNTS.
9.4.8 Concentrados de plaquetas congeladas.
9.4.8.1 Para la congelación de plaquetas deberán emplearse unidades obtenidas por aféresis que se encuentren en el lapso de las primeras 24 horas después de su extracción.
9.4.8.2 Para la crioprotección de las plaquetas se podrá emplear la técnica de dimetilsulfóxido (6% peso/volumen) o la de glicerol a bajas concentraciones (5% peso/volumen).
9.4.8.3 La vigencia y temperatura de conservación de las unidades de plaquetas congeladas se señala la Tabla 16 de este proyecto de Norma.
Tabla 16
Vigencia y conservación de unidades de plaquetas congeladas
Tipo de unidad |
Temperatura de conservación |
Vigencia |
Plaquetas obtenidas por aféresis |
Igual o inferior a -150º C |
Máximo 24 congelación meses A partir de la |
Igual o inferior a -70º C |
Máximo 12 congelación meses A partir de la |
9.4.8.4 Una vez descongeladas y lavadas, las plaquetas se conservarán entre +20º C y +24º C, en agitación suave (véase incisos 9.4.2, 9.4.2.1 y 9.4.2.2 de este proyecto de Norma). Su uso terapéutico será en el menor tiempo posible, sin exceder de 6 horas, de lo contrario, se les deberá dar destino final.
9.4.8.5 Los requisitos de calidad de las unidades de plaquetas descongeladas se señala la Tabla 17 de este proyecto de Norma.
Tabla 17
Requisitos de calidad que deben reunir el 100% de las unidades o mezclas de plaquetas congeladas y descongeladas
Parámetro a verificar |
Especificación |
Cantidad de unidades y frecuencia de verificación |
Volumen |
50 - 200 mL |
Todas las unidades o mezclas preparadas en el mes. |
Cuenta de plaquetas |
> 50% del valor de previo a la congelación |
|
Leucocitos precongelación en unidades leucodepletadas |
<1 x 106 por unidad, en el 90% de las unidades probadas |
|
Control bacteriológico al final del procesamiento. |
Sin desarrollo |
Nota: - En unidades o mezclas leucodepletadas es admisible una pérdida de plaquetas entre el 10 y 15%
- Las unidades de plaquetas descongeladas y reconstituidas son pobres en eritrocitos y leucocitos.
9.5 Concentrado de granulocitos.
9.5.1 Los concentrados de granulocitos para uso terapéutico se deberán obtener mediante aféresis.
9.5.2 Los concentrados de granulocitos se deberán conservar entre +20º y +24º C, preferentemente sin agitación. Su periodo máximo de vigencia será de 24 horas; de no emplearse en este lapso, se les deberá dar destino final.
9.5.3 Los requisitos de calidad que se deberán verificar en todos los concentrados de granulocitos se indican en la Tabla 18 de este proyecto de Norma.
Tabla 18
Requisitos de calidad que deben reunir el 100% de los concentrados de granulocitos
Parámetro a verificar |
Especificación |
Cantidad de unidades y frecuencia de verificación |
HLA |
Tipificación |
Cuando se requiera |
Volumen |
200 a 300 mL |
Todas las unidades |
Cuenta de granulocitos |
>1 x 1010 por unidad |
A cada unidad obtenida |
Control bacteriológico al final del procesamiento |
Sin desarrollo |
Todas las unidades |
9.6 Plasma y crioprecipitados.
9.6.1 Disposiciones comunes:
9.6.1.1 Para obtener plasma fresco y crioprecipitados conservando adecuadamente sus propiedades procoagulantes, se deberá proceder como se indica a continuación:
9.6.1.1.1 Si se obtienen de unidades de sangre fresca, la duración de la flebotomía no habrá excedido de 15 minutos;
9.6.1.1.2 El intervalo entre la extracción de la sangre total de la que se obtendrá el plasma o la plasmaféresis y el inicio de la congelación, preferentemente será de 6 horas, sin exceder nunca de 18 horas. En ese periodo las unidades se habrán mantenido entre +2° C y +10° C;
9.6.1.1.3 La centrifugación para separar el plasma de la sangre total deberá ser a una velocidad de 5,000 g, y
9.6.1.1.4 La velocidad óptima del congelamiento es cuando la temperatura del centro de la unidad de plasma se reduce a -30º C o inferior en un lapso que no exceda de 60 minutos. Para incrementar la velocidad de congelamiento completo, se deberá observar lo siguiente:
9.6.1.1.5 Las bolsas con plasma deberán colocarse de forma que la mayor parte de su superficie reciba la máxima exposición al proceso de enfriamiento, y
Las bolsas deberán someterse a ambientes con temperaturas de -70º C o inferiores. De emplearse ambientes líquidos, se deberán utilizar bolsas en las que se haya demostrado que no sufren daño o que no son penetradas por el disolvente.
Para verificar el logro del objetivo a que se refiere el primer párrafo del inciso 9.6.1.1.4 de este Proyecto de Norma, se deberá contar con método validado para el efecto.
9.6.1.2 No deberán identificarse como plasmas frescos ni se obtendrá de ellos crioprecipitados si no cumplen los requisitos de procesamiento que señala el inciso anterior.
9.6.1.3 La vigencia que deberá asignarse a las unidades de plasma y crioprecipitados se indica la Tabla 19, siempre y cuando se mantengan constantemente a las temperaturas que señala la misma tabla. El periodo de vigencia de las unidades de plasma y los crioprecipitados se cuenta a partir de la extracción de la sangre o de la plasmaféresis. El periodo de vigencia del plasma desprovisto de factores lábiles se cuenta a partir del evento que lo hace considerarlo como tal según la Tabla 19 de este proyecto de Norma. Los requisitos de calidad que deberán reunir el 100% de plasma fresco probados se expresan en la Tabla 20 de este proyecto de Norma.
Tabla 19
Conservación y vigencia de las unidades de plasma y crioprecipitados
Unidad |
Temperatura de conservación |
Intervalos máximos de vigencia (véase nota) |
Plasma fresco, plasma desprovisto de factores lábiles de la coagulación y crioprecipitados |
-30º C o inferior |
36 meses |
-18º C a -30º C |
Tres meses |
|
Plasma fresco descongelados y crioprecipitados descongelados |
+2º C a +6º C |
Seis horas |
Tabla 20
Requisitos de calidad que deberán reunir el 100% de las unidades de plasma fresco probadas
Parámetro a verificar |
Requisitos de calidad (especificación) |
Frecuencia de control |
Volumen |
El establecido ± 10% |
Todas las unidades |
Contenido de proteína total |
Mínimo 50 g/L |
Todas las unidades |
9.6.1.4 Los plasmas que fuesen a destinarse para elaborar hemoderivados deberán conservarse bajo los criterios que indique de la Farmacopea de los Estados Unidos Mexicanos (FEUM, y ediciones actualizadas posteriores).
9.6.1.4.1 Si se mezclan 2 o más unidades antes de la congelación, las operaciones se realizan utilizando dispositivos de conexión estériles o en condiciones asépticas y utilizando recipientes que no hayan sido utilizados previamente.
9.6.1.4.2 Cuando se obtiene por plasmaféresis o de sangre total, el plasma destinado a la recuperación de proteínas lábiles se debe congelar dentro de las 24 h posteriores a la recolección mediante congelación rápido en condiciones validadas para asegurar que se tiene una temperatura de -25°C o menor en el centro de cada unidad de plasma dentro de las 12 h posteriores a su colocación en el equipo de congelación.
9.6.1.4.3 Cuando se obtiene por plasmaféresis, el plasma destinado únicamente a la recuperación de proteínas que no son lábiles en el plasma se congela rápidamente en una temperatura de -20 °C o menor dentro de las 24 h posteriores a la recolección.
9.6.1.4.4 Cuando se obtiene de sangre total, el plasma destinado únicamente a la recuperación de proteínas que no son lábiles, el plasma se separa de los elementos celulares y se congela a una temperatura de -20 °C o menor dentro de las 72 h posteriores a la recolección.
9.6.1.4.5 El plasma congelado se almacena y transporta en condiciones diseñadas y validadas para mantener la temperatura igual o por debajo de -20 °C; si por motivos accidentales, la temperatura de almacenamiento se eleva por encima de -20 °C en una o más ocasiones durante el almacenamiento o el transporte, el plasma se considera apto para el fraccionamiento si se cumplen todas las condiciones siguientes:
9.6.1.4.5.1 El período total de tiempo durante el cual la temperatura supera los -20 ° C no debe ser mayor a las 72 h;
9.6.1.4.5.2 La temperatura no debe superar los -15 °C en más de una ocasión;
9.6.1.4.5.3 La temperatura en ningún momento debe superar los -5 °C.
9.6.1.5 Las bajas temperaturas a partir de -18º C, pueden causar fractura de las bolsas contenedoras del plasma o de los crioprecipitados, por lo que durante el descongelamiento se revisará la existencia de fugas; de haber alguna, se le dará destino final a la unidad.
Las unidades de plasma o crioprecipitados una vez descongeladas no deberán congelarse nuevamente para futuros usos transfusionales, salvo en el caso del plasma sobrenadante tras separar el crioprecipitado.
9.6.2 Plasma.
9.6.2.1 La utilidad más importante del plasma es en el procesamiento de unidades, entre otras, preparación de crioprecipitados y reconstitución de productos sanguíneos celulares. El uso transfusional es limitado. Cuando está adecuadamente prescrito, aproximadamente el 12% de la producción de plasma tiene indicación terapéutica.
Los plasmas no empleados en transfusión se podrán utilizar para la elaboración de hemoderivados.
9.6.2.2 Con el fin de prevenir el daño pulmonar agudo asociado a transfusión, a los plasmas recuperados provenientes de donantes con antecedentes de aloinmunización por causas tales como embarazo o transfusiones previas, no se emplearán con fines transfusionales, sin embargo, podrán reservarse para la elaboración de hemoderivados, o bien, se les dará destino final.
9.6.2.3 El plasma se podrá obtener por procesamiento de sangre fresca de una donación única o mediante aféresis automatizada.
9.6.2.4 Los siguientes se deberán considerar plasmas desprovistos de factores lábiles de la coagulación:
9.6.2.4.1 Los que se hubieran utilizado para obtener crioprecipitados;
9.6.2.4.2 Los que no se hubieran procesado bajo las condiciones que señala el inciso 9.6.1.1 de este proyecto de Norma, así como:
9.6.2.4.3 El que se hubiese obtenido por sedimentación de unidades de sangre. A este plasma se le deberá dar destino final, y
9.6.2.4.4 El plasma fresco conservado adecuadamente (véase tabla 21 de este Proyecto de Norma), que llegó a su límite de vigencia y los plasmas inadecuadamente conservados.
9.6.2.5 Los plasmas frescos que hubieran llegado a su límite de vigencia se clasificarán como plasmas desprovistos de factores lábiles y se le reasignará la vigencia correspondiente. A los crioprecipitados que hubiesen llegado a su límite de vigencia se les dará destino final.
9.6.2.6 Los requisitos de calidad del plasma fresco y del plasma desprovisto de factores lábiles se indican en las Tablas 21 y 22 de este proyecto de Norma, respectivamente.
Tabla 21
Requisitos de calidad que deberán reunir el 100% de las unidades de plasma fresco probadas
Parámetro a verificar |
Requisitos de calidad (especificación) |
Frecuencia del control |
Inspección visual |
a) Integridad de la bolsa: Sin fugas al comprimir la bolsa en un extractor de plasma, antes y después de su congelamiento, y b) Sin color anormal ni restos de fibrina visibles. |
Todas las unidades |
Factor VIIIc |
Minino de 0,7 UI/mL, por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Conteo de células residuales previo al congelamiento |
- Eritrocitos: <6.0 × 109 /L - Leucocitos: <0.1 × 109 /L - Plaquetas: <50 × 109 /L |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Conteo de células residuales previo al congelamiento si se utiliza filtro de última generación (leucodepletado) |
- <1 x 106 por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Tabla 22
Requisitos de calidad que deberán reunir el 100% de las unidades de plasma fresco con reducción de patógenos
Parámetro a verificar |
Requisitos de calidad (especificación) |
Frecuencia del control |
Inspección visual |
a) Integridad de la bolsa: Sin fugas al comprimir la bolsa en un extractor plasmático, antes y después de su congelamiento, y b) Sin color anormal ni coágulos visibles |
Todas las unidades |
Volumen |
El establecido ± 10% |
Todas las unidades |
Fibrinógeno |
Promedio después del proceso de inactivación: ≥60% del porcentaje de la unidad de plasma recién extraido. |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Factor VIIIc |
Minino de 0,7 UI/mL, por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Contenido de proteína total |
Mínimo un límite de 50 g/L |
Todas las unidades |
Inactivación de patógenos |
La concentración residual de las sustancias de inactivación, se reduce a un nivel que no represente un riesgo para los pacientes a los que se aplicará la preparación o interferencia en la prueba de titulación de anticuerpos. |
Todas las unidades |
CEP* Control estadístico de proceso.
9.6.2.7 Podrán lograrse cifras más bajas de leucocitos residuales si en el procesamiento de las unidades de plasma se incluye algún método de leucodepleción específico.
9.6.2.8 Para utilizar los plasmas congelados, frescos o no, deberán descongelarse a temperaturas entre +30° y +37° C mediante técnicas o equipos específicos validados para el efecto, que no afecten, en su caso, los factores lábiles de la coagulación. De emplearse baño de María deberá evitarse que se contamine el puerto de entrada de la bolsa contenedora. Una vez descongelados deberán transfundirse a la brevedad, o bien, conservarse entre +2° y +6° C por un lapso que no exceda de 6 horas con el fin de evitar la pérdida de la actividad de los factores lábiles de la coagulación. De no emplearse para transfusión se les dará destino final.
9.6.3 Crioprecipitados.
9.6.3.1 El crioprecipitado se obtiene por procesamiento del plasma fresco congelado. Adecuadamente procesado y conservado, contiene factor VIII de la coagulación, factor von Willebrand, fibrinógeno, factor XIII y fibronectina.
9.6.3.2 Para utilizar los crioprecipitados deberán descongelarse a temperaturas entre +30° y +37° C mediante técnicas o equipos específicos validados para el efecto, que no afecten los factores lábiles de la coagulación. De emplearse baño de María deberá evitarse que se contamine el puerto de entrada de la bolsa contenedora. Una vez descongelados deberá favorecerse la disolución del producto mediante manipulación suave y transfundirse a la brevedad, o bien, conservarse entre +2° y +6° C por un lapso que no exceda de 6 horas con el fin de evitar la pérdida de sus propiedades procoagulantes, de no transfundirse en ese lapso se le dará destino final.
9.6.3.3 Los requisitos de calidad de los crioprecipitados se indican la Tabla 23 de este proyecto de Norma.
Tabla 23
Requisitos de calidad que deben reunir el 100% de las unidades y mezclas de crioprecipitado probadas
Parámetro a verificar |
Requisitos de calidad (especificación) |
Frecuencia del control |
Inspección visual |
a) Integridad de la bolsa. Sin fugas al comprimir la bolsa en un extractor plasmático antes y después de su congelamiento, y b) Sin color anormal ni coágulos visibles |
Cada día de procesamiento, a todas las unidades |
Volumen |
30 a 40 mL |
Cada día de procesamiento, a todas las unidades o mezclas |
Parámetro a verificar |
Requisitos de calidad (especificación) |
Frecuencia del control |
Factor VIIIc |
> 70 UI por unidad |
Cada dos meses solo en caso de que se utilice en pacientes hemofílicos: a) Mezcla de seis unidades de distintos grupos sanguíneos al primer mes de almacenamiento, y b) Mezcla de seis unidades de distintos grupos sanguíneos del último mes de vigencia. |
Fibrinógeno |
> 140 mg por unidad |
Calcular la frecuencia de verificación con base en la producción mensual y obtenida por CEP* |
Factor von Willebrand |
> 100 IU por unidad |
Cada dos meses solo en caso de que se utilice en pacientes con enfermedad de von Willebrand: a) Mezcla de seis unidades de distintos grupos sanguíneos durante el primer mes de almacenamiento, y b) Mezcla de seis unidades de distintos grupos sanguíneos durante el último mes de vigencia. |
9.6.3.4 Los requisitos de calidad para las mezclas de crioprecipitados relativos al volumen y contenidos de factor VIII coagulante, fibrinógeno y factor von Willebrand, serán iguales a la resultante de multiplicar los valores obtenidos correspondientes por el número de unidades que integran la mezcla.
9.7 Inactivación en productos sanguíneos.
A criterio del responsable sanitario del Banco de Sangre, los productos sanguíneos que vayan a destinarse para uso transfusional, podrán someterse a técnicas in-vitro validadas y estandarizadas que impidan la proliferación de agentes potencialmente infectantes o de células inmunocompetentes, mediante métodos como inactivación fotodinámica, fotoquímica, solvente detergente u otros que permitan el mantenimiento de propiedades terapéuticas, en su caso, de su viabilidad y que no provoquen toxicidad en el receptor. Estos métodos no sustituyen la irradiación de productos sanguíneos para la prevención de la enfermedad injerto contra huésped (u hospedero) a no ser que el método de inactivación permita que los linfocitos sean inactivados por el proceso, procedimiento que deberá estar validado por el responsable sanitario del servicio de sangre que desee emplearlo en sustitución de la irradiación.
Las técnicas de inactivación viral para el plasma fresco congelado, podrán llevarse a cabo en el Banco de Sangre o por manufactura en la industria farmacéutica. Los plasmas que hayan sido inactivados deberán tener las siguientes consideraciones:
9.7.1 Volumen: de acuerdo con el sistema utilizado;
9.7.2 Factor VIIIc al final por unidad mínimo de 0,7 UI/mL;
9.7.3 Fibrinógeno al final por unidad será: ≥ 140 mg;
9.7.4 Proteínas totales como mínimo un límite de 50 g/L;
9.7.5 La concentración residual de las sustancias de inactivación se reduce a un nivel que no represente un riesgo para los pacientes a los que se aplicará la preparación o interferencia en la prueba de titulación de anticuerpos.
9.7.6 Las indicaciones de la Tabla 24 de este proyecto de Norma.
Tabla 24
Técnicas para la reducción de patógenos en los hemocomponentes a transfundir
Producto sanguíneo |
Método fotodinámico |
Método fotoquímico |
Otro método |
Plasma: unidades individuales |
Riboflavina (vitamina B2) + Luz UV |
Amotosaleno (psoraleno) + Luz UV |
Tratamiento con Solvente/detergente |
Azul de metileno + Luz |
|||
Concentrado plaquetario |
Riboflavina (vitamina B2) + Luz UV |
Amotosaleno (psoraleno) + Luz UV |
*UV:Ultravioleta
9.8 Disposiciones para la irradiación de productos sanguíneos.
Para reducir el riesgo de enfermedad injerto contra huésped (u hospedero) en receptores susceptibles (véase inciso 12.4 de este proyecto de Norma), se deberán irradiar las unidades celulares que pretendan transfundirse, de conformidad con lo siguiente:
9.8.1 Se sugiere utilizar dispositivos de irradiación de rayos X, como alternativa a la radiación gamma de los radionúclidos de Cesio (Cs) 137 y Cobalto (Co) 60. En caso de aún contar con fuentes de Cs 137 y Co 60, se tendrá en consideración la seguridad e higiene del ambiente y el personal según los estándares del manual de "Identificación de fuentes y dispositivos radioactivos" del Organismo Internacional de Energía Atómica (OIEA) y de conformidad con la NORMA Oficial Mexicana NOM-012-STPS-2012. Condiciones de seguridad y salud en los centros de trabajo donde se manejen fuentes de radiación ionizante.
9.8.2 Los equipos y los procedimientos de irradiación deberán ser validados y verificados de acuerdo con la vida media del radionúclido con el fin de determinar el tiempo necesario de irradiación de los productos sanguíneos y asegurar que reciban la dosis necesaria. En caso de utilizar otros equipos emisores de radiación ionizante será necesario desarrollar protocolos de validación y verificación equivalentes, que garanticen que la irradiación se realiza correcta y homogéneamente;
9.8.3 Los equipos emisores de radiación ionizante deberán recibir anualmente mantenimiento preventivo y, en su caso, correctivo cuando por el mismo uso y desgaste de los sistemas del equipo se vean afectadas;
9.8.4 Mensualmente, deberá efectuarse dosimetría personal. La dosimetría ambienta deberá realizarse de conformidad a lo señalado por la Licencia de Operación y el Reglamento General de Seguridad Radiológica;
9.8.5 El tiempo de exposición deberá ajustarse para asegurar que los productos sanguíneos reciban una dosis no menor de 2,500 cGy (dosis mínima) y sin que alguna parte de las unidades reciban más de 5,000 cGy (dosis máxima);
9.8.6 Deberá verificarse la eficiencia de la irradiación empleando cintas reactivas sensibilizadas para cada lote de unidades irradiadas o bien, por medio de cultivo mixto de linfocitos, mensualmente, en el 1% o 4 unidades lo que sea mayor;
9.8.7 Las unidades de concentrados de eritrocitos que vayan a irradiarse no deberán tener más de 14 días después de su extracción. Los concentrados o mezclas de plaquetas podrán radiarse en cualquier momento dentro de su periodo de vigencia. Las unidades de granulocitos se irradiarán tan pronto como sea posible después de haberse preparado;
9.8.8 No será necesario irradiar los componentes acelulares que hubiesen estado congelados;
9.8.9 Los concentrados de eritrocitos o de plaquetas irradiados que fuesen a usarse en transfusión intrauterina o exsanguineotransfusión en neonatos, deberán transfundirse dentro de las 48 horas siguientes a la irradiación;
9.8.10 Las unidades de concentrados de eritrocitos irradiados tendrán una vigencia máxima de 14 días a partir de la irradiación, si el anticoagulante usado así lo permite. Los concentrados de plaquetas y granulocitos irradiados mantendrán su periodo de vigencia asignado, y
9.8.11 Las unidades irradiadas podrán emplearse en cualquier receptor.
9.8.12 El encargado de seguridad radiológica del establecimiento deberá observar las demás disposiciones que señala la Licencia de Operación y el Reglamento General de Seguridad Radiológica.
9.8.13 Se podrán emplear alternativamente a la irradiación, tecnologías validadas y verificadas en eficiencia y eficacia, en sustitución de la misma, siempre que el responsable sanitario cuente con la evidencia documental de la utilidad en la inactivación de leucocitos.
10 Determinaciones analíticas
10.1 Para la realización de las pruebas de detección de agentes infecciosos transmisibles por transfusión y las pruebas de inmunohematología, el personal asignado por el responsable sanitario del Banco de Sangre, del Servicio de Transfusión Hospitalario, el Centro de Procesamiento o en su caso del Centro de Calificación Biológica deberá observar lo siguiente:
10.1.1 El personal deberá recibir capacitación y adiestramiento adecuada para el desarrollo de las pruebas y tendrá constancia de ello;
10.1.2 Únicamente se emplearán reactivos validados que cuenten con número de registro sanitario de la Secretaría y tengan una caducidad vigente, y
10.1.3 Las pruebas se realizarán de manera uniforme, siguiendo buenas prácticas de laboratorio y los Procedimientos Normalizados de Operación desarrollados para tal fin y las recomendaciones e instrucciones proporcionadas por el fabricante de los reactivos.
10.1.4 El responsable sanitario del servicio de sangre, deberá implementar las medidas de bioseguridad necesarias para asegurar que el personal minimice su exposición a agentes biológicos infecciosos.
10.1.5 Los equipos e instrumentos que se utilicen para realizarlas deberán ser calibrados y verificados y contar con registros de los mismos.
10.2 Los laboratorios de los Bancos de Sangre, Centros de Procesamiento, Centros de Calificación Biológica y, en su caso, los Servicios de Transfusión Hospitalario deberán contar con los Procedimientos Normalizados de Operación escritos para el manejo, trasvasado y etiquetado de las muestras que permitan garantizar su correcta trazabilidad desde la fase preanalítica hasta la postanalítica.
10.3 Los Bancos de Sangre, Centros de Procesamiento, Centros de Calificación Biológica y, en su caso, los Servicios de Transfusión Hospitalario, deberán tener y conservar registros de todas las determinaciones analíticas que efectúen en donantes y receptores. Los registros garantizarán la identificación inequívoca de las muestras de los donantes o receptores. Los resultados de las determinaciones analíticas serán notificados a la brevedad al personal que suministre las unidades para su uso, traslado a otro establecimiento o transfusión.
10.4 Pruebas para la detección de agentes infecciosos transmisibles por transfusión.
10.4.1 Con las muestras sanguíneas tomadas en cada donación de sangre y productos sanguíneos durante el inicio de la flebotomía, se deberán efectuar las pruebas para la detección de agentes transmisibles por transfusión invariablemente antes de la liberación para uso terapéutico. Estas pruebas deberán realizarse en toda donación independientemente de que antes de efectuar las pruebas se hubiese dado destino final al producto sanguíneo de que se trate.
10.4.2 Las pruebas para la detección de los agentes infecciosos transmisibles por transfusión deberán incluir obligatoriamente la detección de antígenos, anticuerpos, ADN y/o ARN de los siguientes:
10.4.2.1 Treponema pallidum;
10.4.2.2 Virus B de la hepatitis (antígeno de superficie y DNA);
10.4.2.3 Virus C de la hepatitis (anticuerpos y ARN);
10.4.2.4 Virus de la inmunodeficiencia humana tipos 1 y 2, (antígeno p24, anticuerpos y ARN) y
10.4.2.5 Trypanosoma cruzi.
10.4.3 Cuando por la situación epidemiológica de la región geográfica donde se encuentra el establecimiento o de la de procedencia del donante, sus antecedentes personales o sus factores de riesgo para adquirir infecciones, o bien, por las características de los futuros receptores y su susceptibilidad a adquirir o desarrollar enfermedad, el Servicio de Sangre deberá efectuar y documentar pruebas adicionales para la detección de los agentes infecciosos transmisibles por transfusión. Las pruebas adicionales podrán incluir la detección de los agentes siguientes:
10.4.3.1 Brucella;
10.4.3.2 Plasmodium;
10.4.3.3 Toxoplasma;
10.4.3.4 Retrovirus HTLV tipos I y II;
10.4.3.5 Virus E de la Hepatitis y,
10.4.3.6 Otros agentes.
10.4.4 Para que el Banco de Sangre autorice el uso terapéutico de las unidades de sangre y productos sanguíneos, deberá contar con resultados inequívocamente negativos en las pruebas de detección de agentes transmisibles.
10.4.5 En procedimientos de laboratorio correctamente efectuados y que su control de calidad demuestre que los resultados son confiables en un porcentaje ≥ 95%, si el resultado de la prueba de tamizaje es reactivo, el personal que realiza las pruebas, deberá proceder como señalan las Figuras 1, 2 y 3 de este proyecto de Norma.
10.4.6 Las pruebas de confirmación y/o suplementarias, deberán realizarse preferentemente por los Laboratorios Estatales de Salud Pública y/o por el Instituto de Diagnóstico y Referencia Epidemiológicos, cuando no puedan llevarse a cabo en el Servicio de Sangre.
10.4.7 La responsabilidad de notificación de caso positivo confirmado por laboratorio a la jurisdicción sanitaria, será del banco de sangre, una vez que se cuente con el resultado de la prueba confirmatoria o suplementaria. De cualquier forma, se deberá dar aviso a la DGE por escrito y conservar la evidencia documental.
Figura 1
Diagrama de flujo para el tamizaje de sífilis en donantes de sangre
Figura 2
Diagrama de flujo para el tamizaje de T. cruzi en donantes de sangre
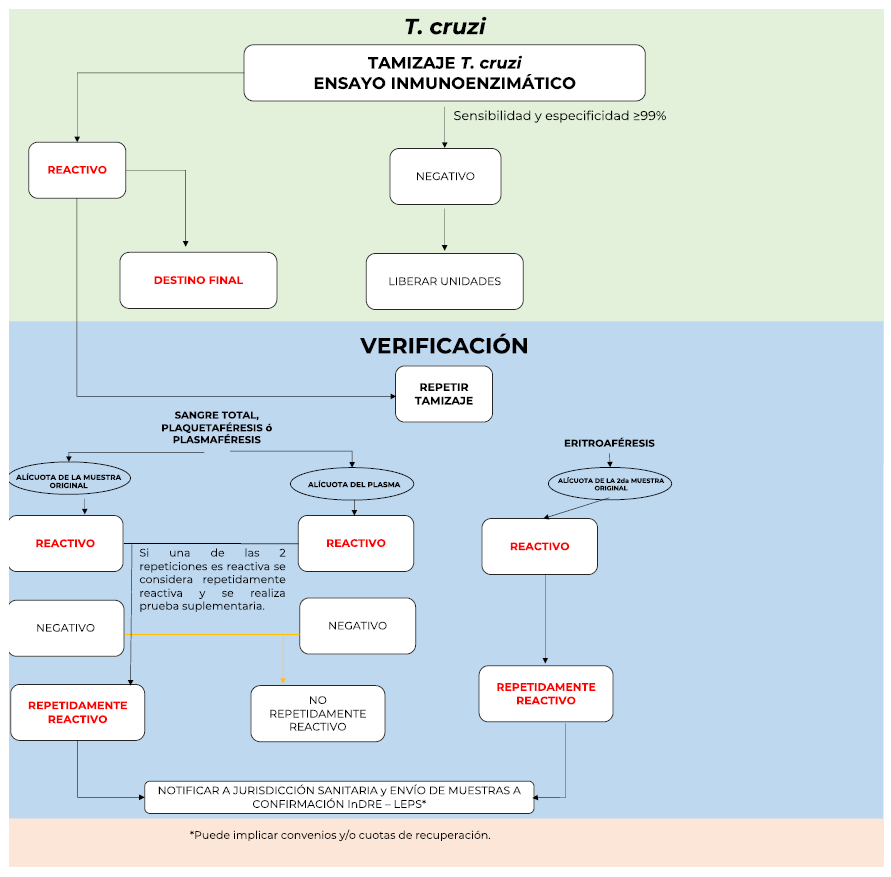
Figura 3
Diagrama de flujo para el tamizaje de VIH, VHB y VHC por medio de NAT
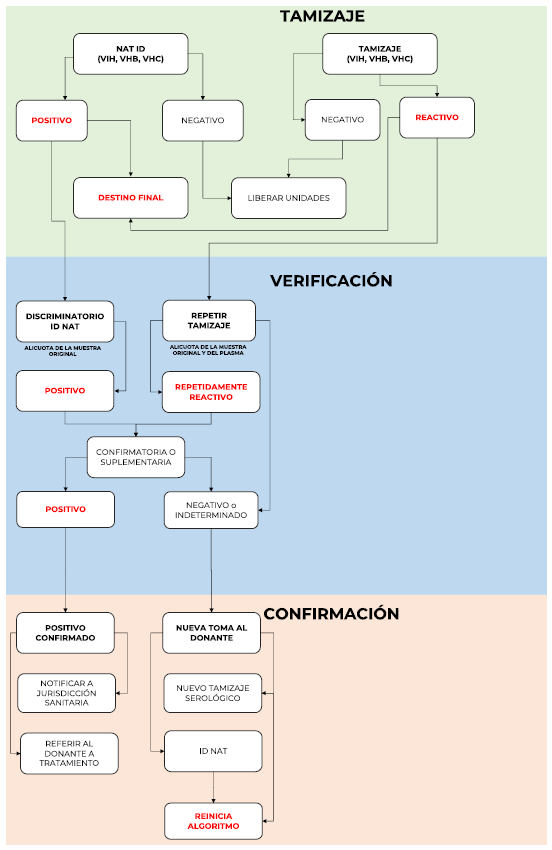
10.4.8 De haber discrepancia entre los resultados de la muestra original y de la alícuota tomada de la unidad, no deberá liberarse el lote de unidades estudiadas hasta haber identificado la unidad reactiva.
10.4.9 Con las muestras sanguíneas que resultaron repetidamente reactivas o con nuevas muestras del donante tomadas con posterioridad, el establecimiento deberá efectuar las pruebas confirmatorias o, en su caso, suplementarias o bien, si no cuenta con la capacidad técnica, referir a la brevedad las muestras a los Laboratorios Estatales de Salud Pública o al el Instituto de Diagnóstico y Referencia Epidemiológicos, otro Banco de Sangre, Centros de Calificación Biológica o a laboratorios con capacidad técnica suficiente y comprobada.
10.4.10 Para la notificación a la Secretaría de las anomalías detectadas en las pruebas de laboratorio se deberá proceder como señala el inciso 19.8 de este proyecto de Norma.
10.4.11 Detección de Treponema pallidum.
10.4.11.1 Tamizaje. Se deberá realizar mediante cualquiera de las pruebas siguientes:
10.4.11.2 Preferentemente por medio de identificación de anticuerpos específicos mediante pruebas treponémicas con especificidad ≥ 98.50%, tales como:
10.4.11.3 Inmunocromatografía;
10.4.11.4 Ensayo inmunoenzimático, u
10.4.11.5 Otras con sensibilidad y especificidad igual o mayor.
10.4.11.6 Identificación de reaginas mediante una prueba de aglutinación de partículas, entre las siguientes:
10.4.11.7 VDRL, o
10.4.11.8 RPR, o
10.4.11.9 El control de calidad de la prueba de tamizaje efectuado en cada corrida se demuestra cuando la prueba tiene una sensibilidad suficiente para detectar un control positivo débil.
10.4.11.10 Confirmatoria. Se deberán emplear pruebas treponémicas que tengan especificidad mínima 99 %, entre otras, cualquiera de las siguientes:
10.4.11.11 Hemaglutinación contra Treponema;
10.4.11.12 Anticuerpos fluorescentes contra el Treponema;
10.4.11.13 Inmunofluorescencia indirecta;
10.4.11.14 Inmovilización del treponema, u
10.4.11.15 Otras con especificidad igual o mayor.
10.4.11.16 La prueba se validará a través de los controles y especificaciones que señale el fabricante. Los resultados de la prueba serán confiables siempre y cuando sean los esperados y señalados por el fabricante.
10.4.12 Detección del Virus B de la hepatitis
10.4.12.1 Tamizaje. Se deberá realizar mediante pruebas de detección del antígeno de superficie del virus B de la hepatitis, con pruebas que tengan una sensibilidad ≥ 99.5 % y especificidad ≥ 99.0 %, tales como:
10.4.12.2 Ensayo inmunoenzimático;
10.4.12.3 Inmunoensayo por quimioluminiscencia, y
10.4.12.4 Otras con sensibilidad y especificidad igual o mayor.
10.4.12.5 El control de calidad de la prueba de tamizaje efectuado en cada corrida se demuestra cuando la prueba tiene una sensibilidad suficiente para detectar un control positivo débil.
10.4.12.6 Confirmatoria. Se deberán realizar mediante la detección de antígenos con una prueba de neutralización con anticuerpos con especificidad ≥ 99.5%.
10.4.12.7 La prueba se validará a través de los controles y especificaciones que señale el fabricante.
10.4.13 Detección del virus C de la hepatitis
10.4.13.1 Tamizaje. Se deberá realizar mediante pruebas de detección de anticuerpos contra el virus C o detección simultanea de antígenos virales y anticuerpos contra el virus que tengan una sensibilidad ≥ 99.5% y especificidad ≥ 99%, entre las siguientes:
10.4.13.2 Ensayo inmunoenzimático;
10.4.13.3 Inmunoensayo por quimioluminiscencia, y
10.4.13.4 Otras con sensibilidad y especificidad igual o mayor.
10.4.13.5 El control de calidad de la prueba de tamizaje efectuado en cada corrida se demuestra cuando la prueba tiene una sensibilidad suficiente para detectar un control positivo débil.
10.4.13.6 Confirmatoria. Se deberá realizar mediante la prueba de "inmunoblot" recombinante u otras con sensibilidad y especificidad igual o mayor. Para identificación de agentes virales infecciosos será por NAT.
10.4.13.7 La prueba se validará a través de los controles y especificaciones que señale el fabricante.
10.4.14 Detección del virus de la inmunodeficiencia humana, tipos 1 y 2.
10.4.14.1 Tamizaje. Se deberá realizar mediante pruebas de marcadores de infección del virus que tengan una sensibilidad ≥ 99.5% y una especificidad ≥ 99%, entre las siguientes:
10.4.14.2 Ensayo inmunoenzimático combinado para la determinación de antígenos y anticuerpos virales;
10.4.14.3 Ensayo inmunoenzimático;
10.4.14.4 Inmunoensayo por quimioluminiscencia, y
10.4.14.5 Otras que tengan sensibilidad y especificidad igual o mayor.
10.4.14.6 El control de calidad de la prueba de tamizaje efectuado en cada en cada corrida, se demuestra cuando la prueba tiene una sensibilidad suficiente para detectar un control positivo débil.
10.4.14.7 Confirmatoria. La confirmación se deberá realizar mediante una prueba de detección de anticuerpos contra el VIH tipos 1 y 2, entre cualquiera de las siguientes:
10.4.14.8 Prueba inmunocromatográfica de un solo uso;
10.4.14.9 Carga viral;
10.4.14.10 Inmunofluorescencia;
10.4.14.11 Inmunoensayo recombinante, u
10.4.14.12 Otras metodologías más avanzadas.
10.4.14.13 La prueba se validará a través de los controles y especificaciones que señale el fabricante.
10.4.15 Detección del Trypanosoma cruzi
10.4.15.1 Tamizaje. Se deberá realizar mediante pruebas de detección de anticuerpos contra el
10.4.15.2 Trypanosoma cruzi que tengan una sensibilidad y especificidad ≥ 99%, entre las siguientes:
10.4.15.3 Ensayo inmunoenzimático;
10.4.15.4 Otras con especificidad y sensibilidad igual o mayor.
10.4.15.5 El control de calidad de la prueba de tamizaje efectuado en cada corrida se demuestra cuando la prueba tiene una sensibilidad suficiente para detectar un control positivo débil.
10.4.15.6 Prueba suplementaria. Se deberá emplear una prueba de detección de anticuerpos contra el Trypanosoma cruzi, que tenga un formato distinto a la prueba empleada para el tamizaje y que tenga una especificidad superior.
10.4.15.7 La prueba se validará a través de los controles y especificaciones que señale el fabricante.
10.4.16 Pruebas para la detección de agentes transmisibles en condiciones especiales
10.4.16.1 Detección de Brucella. Las pruebas para la detección de Brucella que se indican a continuación, se practicarán a las personas consideradas de riesgo, tales como: las que ingieren productos lácteos no pasteurizados, las que trabajan con animales domesticados, especialmente ganado vacuno, caprino, porcino, ovejas, ciervos, alces o que tienen contacto con sus carnes, excreciones, secreciones, placentas o sus cadáveres, así como las que trabajan en granjas, mataderos, curtidurías y los veterinarios:
10.4.16.1.1 Pruebas de tamizaje. Se efectuará mediante pruebas de aglutinación en placa con antígeno teñido con rosa de bengala o ensayo inmunoenzimático para la detección de anticuerpos totales o de tipo IgM, y
10.4.16.1.2 Prueba confirmatoria. Se realizará a través de metodologías como titulación de anticuerpos mediante aglutinación en presencia de 2 mercaptoetanol y otras disponibles para el efecto.
10.4.16.2 Detección de Plasmodium. Las pruebas que se indican a continuación se practicarán para definir la aceptabilidad de un donante que se encuentre en cualquiera de las condiciones que señala la Tabla 3 de este proyecto de Norma.
10.4.16.2.1 Ensayo inmunoenzimático;
10.4.16.2.2 Inmunofluorescencia, y
10.4.16.2.3 Investigación del parásito con microtubo con naranja de acridina.
10.4.16.2.4 Otras con especificidad y sensibilidad igual o mayor.
10.4.16.2.5 Detección de citomegalovirus. Se emplearán pruebas de detección de anticuerpos tipo inmunoglobulina M contra el citomegalovirus, entre otras, con cualquiera de las técnicas siguientes:
10.4.16.2.5.1 Ensayo inmunoenzimático, y
10.4.16.2.5.2 Inmunofluorescencia.
10.4.16.3 Detección de Toxoplasma gondii. Se emplearán pruebas de detección de anticuerpos mediante ensayo inmunoenzimático.
10.4.16.4 Retrovirus HTLV I y II:
10.4.16.4.1 Tamizaje. Detección de anticuerpos mediante pruebas que tengan una sensibilidad y especificidad ≥ 95%, entre otras:
10.4.16.4.2 Ensayo inmunoenzimático;
10.4.16.4.3 Amplificación de ácidos nucleicos;
10.4.16.4.4 Otras con especificidad y sensibilidad igual o mayor, y
10.4.16.4.5 Confirmatoria. Detección de anticuerpos por inmunoblot o bien por detección de genoma viral mediante amplificación de ácidos nucleicos.
10.4.16.5 Virus E de la Hepatitis:
10.4.16.5.1 Tamizaje. Detección de anticuerpos mediante ensayo inmunoenzimático que tenga una sensibilidad y especificidad ≥ 99%, entre otras:
10.4.16.5.2 Amplificación de ácidos nucleicos, y
10.4.16.5.3 Otras con especificidad y sensibilidad igual o mayor, y
10.4.16.5.4 Confirmatoria. Detección de anticuerpos por inmunoblot o bien por detección de genoma viral mediante amplificación de ácidos nucleicos.
10.4.17 Los Bancos de sangre, centros de calificación biológica y centros de procesamiento deberán realizar pruebas de amplificación de ácidos nucleicos, para VIH, Virus B de la hepatitis y virus C de la hepatitis, mediante técnicas de amplificación mediada por transcripción, de reacción en cadena de la polimerasa, o equivalentes, y deberán observar lo siguiente:
10.4.18 Estas pruebas no sustituyen a las pruebas de detección de agentes virales transmisibles por transfusión a que se refiere este capítulo, ni son útiles como pruebas confirmatorias, y
10.4.19 Deberán efectuarse y verificarse de conformidad con los estándares de la Organización Mundial de la Salud (OMS) y las instrucciones del fabricante.
10.5 Hemoclasificación y hemocompatibilidad.
10.5.1 Disposiciones comunes:
10.5.1.1 Los Bancos de Sangre, los Servicios de Transfusión Hospitalario deberán realizar las pruebas de compatibilidad sanguínea antes de cada transfusión alogénica, salvo en los casos siguientes:
10.5.1.1.1 Cuando el Banco de Sangre o el Servicio de Transfusión Hospitalario la suministren a otro establecimiento que se responsabilizará en hacerlas, o
10.5.1.1.2 Cuando el establecimiento reciba las unidades con los estudios de compatibilidad previamente realizados.
10.5.1.2 Se realizará rastreo de anticuerpos irregulares a todos los donantes, si resulta positivo, deberá realizarse identificación del o los anticuerpos irregulares. De haber anticuerpos irregulares en la sangre de un donante, se deberá dar destino final al plasma.
10.5.1.3 El responsable del Banco de Sangre o del Servicio de Transfusión Hospitalario, deberá realizar o garantizar que se hayan hecho las pruebas de hemoclasificación y hemocompatibilidad necesarias, de acuerdo con el componente que se fuese a transfundir, antes de cada transfusión de unidades alogénicas.
10.5.1.4 Las pruebas que se emplean para demostrar compatibilidad sanguínea incluyen:
10.5.1.4.1 Hemoclasificación de los sistemas ABO y Rh (antígeno D);
10.5.1.4.2 Investigación (rastreo y eventual identificación) de anticuerpos irregulares de importancia clínica en aquellos receptores, en que su condición clínica así lo requiera, multitransfundidos o con riesgo a hemólisis.
10.5.1.4.3 Pruebas cruzadas.
10.5.1.5 Las pruebas a que hace referencia el inciso anterior se podrán realizar en tubo, soporte sólido, gel o esferas de vidrio. Se podrán utilizar técnicas manuales o automatizadas que previamente hayan sido autorizadas para uso por la Secretaría de Salud.
10.5.1.6 Se deberá procesar un control de calidad de reactivos y técnicas, que asegure los resultados, así como conservar la evidencia de su realización por medios físicos o electrónicos.
10.5.1.7 Para la determinación del antígeno D débil o negativo deberá incluirse la prueba de antiglobulina humana para la confirmación de la positividad o negatividad del antígeno D.
10.5.1.8 Antes de cada transfusión de preparados con eritrocitos se deberán realizar prueba cruzada mayor, independientemente que se hubiera realizado el rastreo e identificación de anticuerpos irregulares.
10.5.1.9 Con el fin de asegurar la confiabilidad en la determinación de grupos ABO y Rh (D), rastreo e identificación de anticuerpos irregulares y pruebas cruzadas, se deberá realizar el control de calidad de los reactivos, equipos e instrumentos de conformidad a lo señalado en el capítulo 16 denominado "Evaluación de la conformidad y control de calidad" de este proyecto de Norma.
10.5.1.10 Los Bancos de sangre o servicios de transfusión hospitalarios que efectúen pruebas de compatibilidad deberán conservar adecuadamente, las muestras sanguíneas del donante y del receptor por un mínimo de siete días contados a partir de la transfusión de la unidad.
Se considera conservación adecuada de las muestras cuando el suero o plasma se almacena a -18º C o inferior y los eritrocitos entre +2º C y +6º C.
10.5.1.11 El establecimiento deberá conservar registros de las acciones realizadas.
10.5.2 Hemoclasificación del grupo ABO.
10.5.2.1 La clasificación del grupo ABO se deberá realizar en todos los donantes y receptores, mediante las pruebas siguientes:
10.5.2.1.1 Pruebas de aglutinación:
10.5.2.1.1.1 Prueba directa: permite identificar la presencia o ausencia de los antígenos A y B en los eritrocitos, mediante el empleo de reactivos hemoclasificadores anti-A y anti-B de origen monoclonal;
10.5.2.1.1.2 Prueba inversa: permite identificar la presencia o ausencia de los anticuerpos regulares anti-A y anti- B en suero o plasma, utilizando eritrocitos con antígeno A1 y B, y
En cada clasificación de grupo ABO, se deberá incluir un control o que valide el resultado
Método de genotipificación sanguínea.
10.5.2.2 No será necesario efectuar la prueba inversa referida en la segunda viñeta del inciso 10.5.2.1.1.2 en los casos siguientes:
10.5.2.2.1 Para ratificar el grupo sanguíneo de los concentrados de eritrocitos ricos en solución aditiva o con muy bajo contenido plasmático;
10.5.2.2.2 En receptores de cuatro meses o menores, o
10.5.2.2.3 Cuando la clasificación del grupo ABO se hubiera realizado mediante genotipificación sanguínea.
10.5.2.3 No se clasificará una unidad o a un receptor en el sistema ABO hasta haber resuelto cualquier discrepancia que hubiese entre la prueba directa y la inversa, utilizando las técnicas disponibles en el laboratorio.
10.5.2.4 En caso de donantes regulares deberá revisarse y compararse la clasificación de grupo ABO que se tenga en los registros de donaciones previas.
10.5.2.5 En receptores recientemente transfundidos y en mujeres con hemorragia feto-materna cuantiosa, en quienes la hemoclasificación se vea dificultada por la presencia de reacciones de campo mixto, se deberá contar con un Procedimiento Normalizado de Operación que establezca el cómo se debe proceder a fin de resolver el problema antes de transfundir.
Se podrán obviar las pruebas a que hace referencia este inciso cuando se identifique el grupo ABO y el antígeno Rh (D) por medio de genotipificación sanguínea a partir del ácido desoxirribonucleico extraído de otra fuente diferente a la de la sangre, por ejemplo, de la saliva.
10.5.3 Hemoclasificación del antígeno Rh (D).
10.5.3.1 La identificación del antígeno D se deberá realizar en los donantes mediante las pruebas siguientes:
10.5.3.1.1 Prueba de aglutinación directa: se emplea un reactivo anti-D de origen monoclonal que contenga inmunoglobulinas M o M+G (mezcla M y G) y que sea diseñado para detectar variantes, que permita identificar la presencia o ausencia del antígeno D en los eritrocitos, y
10.5.3.1.2 Prueba de antiglobulina humana: si la prueba de aglutinación directa referida en el inciso anterior resultase negativa, los glóbulos rojos deberán someterse a una prueba de antiglobulina humana (prueba de Coombs) para demostrar la presencia o ausencia del antígeno D expresado débilmente o sus variantes. Se deberá usar un Anti-D que contenga inmunoglobulina G y que provenga de una clona diferente al Anti-D IgM utilizado para la determinación directa.
Cada una de las pruebas para la identificación del antígeno D, se deberá validar mediante una prueba de control, que permita demostrar la ausencia de aglutinación inespecífica.
Se pueden obviar las pruebas a que hace referencia este inciso cuando se identifique el antígeno D por genotipificación sanguínea.
10.5.3.2 La presencia del antígeno D, expresado débilmente o sus variantes, clasificarán a los eritrocitos como POSITIVOS y su ausencia como NEGATIVOS.
10.5.3.3 La identificación del antígeno D se deberá realizar en los receptores y mujeres gestantes mediante las pruebas siguientes:
10.5.3.3.1 Prueba de aglutinación directa: se emplea un reactivo anti-D de origen monoclonal que contenga inmunoglobulinas M y que no esté diseñado para detectar variantes, que permita identificar la presencia o ausencia del antígeno D en los eritrocitos, y
10.5.3.3.2 Prueba de antiglobulina humana: esta prueba no se realizará para receptores. El paciente receptor se clasificará como Rh (D) positivo o negativo, únicamente con la prueba referida en el inciso anterior.
Cada una de las pruebas para la identificación del antígeno D, se deberá validar mediante una prueba de control, que permita demostrar la ausencia de aglutinación inespecífica.
Se pueden obviar las pruebas a que hace referencia este inciso cuando se identifique el antígeno D por genotipificación sanguínea.
10.5.3.4 El banco de sangre deberá determinar el fenotipo Rh (D) a todos los donantes y pacientes, y deberá formar parte de la información que contenga la etiqueta de la unidad de concentrado eritrocitario, cuando se obtenga este componente sanguíneo, en el caso de donantes; en el caso del paciente, la información se incluirá en el expediente clínico del mismo.
10.5.3.5 Para fines transfusionales, los pacientes se clasificarán como Rh (D) positivo cuando haya presencia del antígeno D y se clasificarán como Rh (D) negativos cuando haya ausencia del antígeno D, según pruebas realizadas en el inciso 10.5.3.2 de este proyecto de norma.
10.5.3.5.1 Los laboratorios de los servicios de sangre que cuenten con serotecas y hematecas deberán cumplir con lo siguiente:
10.5.3.5.1.1 Tener el equipamiento e instalaciones necesarias para poder conservar en congelación sueros y eritrocitos según la técnica de congelación que utilicen.
10.5.3.5.1.2 Deberán tener el soporte científico de cada muestra a guardar en la seroteca y hemateca que compruebe las características de la muestra que la hacen poco frecuente o rara.
10.5.3.5.1.3 Etiquetar perfectamente todos los sueros y eritrocitos que se conservarán con: fecha de preparación, número consecutivo interno de seroteca o hemateca,
10.5.3.5.1.4 Tener una base de datos de cada suero o muestra de eritrocitos que permita conocer las características de los mismos y así poder emplearlos en casos de resolución de casos de inmunohematología.
10.5.3.5.1.5 Cada que el laboratorio o Banco de Sangre emplee un suero de la seroteca o eritrocitos de la hemateca como reactivos, buscará la manera de tener medios de validación de cada corrida, preferentemente usando controles positivos y/o negativos.
10.5.3.5.1.6 Las técnicas en las que se empleen el uso de sueros o eritrocitos de seroteca y hemateca deberán estar previamente validadas y estandarizadas.
10.5.4 Otros sistemas de grupo.
La clasificación de otros sistemas de grupo distintos al ABO y al Rh (D), se deberá realizar mediante pruebas de aglutinación que permitan identificar la presencia o ausencia de los antígenos eritrocitarios de que se trate, empleando reactivos hemoclasificadores específicos o alternativamente a estas pruebas, podrá realizarse genotipificación sanguínea.
10.5.4.1 La fenotipificación de otros antígenos eritrocitarios:
10.5.4.1.1 Se realizará para búsqueda de unidades fenotipo compatible en pacientes con anticuerpos irregulares, se realizará para pacientes y donantes elegibles a prueba cruzada electrónica, se podrá realizar para transfundir unidades fenotipo Rh+Kell compatible o Rh, Kell, Fya, Fyb, Jka, Jkb, S, S y Dia compatible y así prevenir aloinmunización en pacientes mujeres en edad fértil, pacientes con anemia falciforme, pacientes con talasemias, pacientes que requieren transfusiones recurrentes por otros padecimientos.
10.5.4.1.2 La investigación de antígenos eritrocitarios deberá realizarse con antisueros obtenidos de una fuente confiable, mediante técnicas validadas y estandarizadas.
10.5.4.1.3 En casos de investigación de antígenos donde no existen antisueros comerciales, se podrá utilizar un suero de alguna seroteca.
10.5.4.1.4 Se puede predecir el fenotipo a través del genotipo con técnicas validadas de biología molecular.
10.5.4.1.5 Los sueros utilizados deberán cumplir los requisitos de control de calidad como se indica en la Tabla 40 de este proyecto de Norma. El control de calidad se realizará cada día de uso.
10.5.5 Rastreo de anticuerpos irregulares de importancia clínica.
10.5.5.1 El rastreo de anticuerpos irregulares de importancia clínica, se deberá realizar en todos los donantes y receptores con semipaneles de eritrocitos diseñados para detectar anticuerpos de importancia clínica de la población objetivo o similar.
10.5.5.2 El rastreo de anticuerpos irregulares en donadores podrá realizarse con semipaneles como se menciona en el inciso anterior o usando un "pool" de eritrocitos que haya sido diseñado y validado por el fabricante para este uso.
10.5.5.3 En caso de tener rastreo de anticuerpos irregulares positivo en pacientes que van a recibir transfusión se deberá hacer la identificación de los anticuerpos y entregar unidades fenotipo compatible, es decir, que no tengan (carezcan) los antígenos correspondientes a los anticuerpos identificados. En caso de tener un anticuerpo contra antígeno de alta frecuencia o un paciente con mezcla de más de 3 anticuerpos deberá buscarse preferentemente un donante compatible, es decir, un donante que no tenga los antígenos correspondientes a los anticuerpos identificados, en caso de no encontrar un donante compatible se deberá valorar la urgencia de la transfusión con el equipo médico y de Banco de Sangre, optar por otras medidas terapéuticas o buscar otra solución factible.
10.5.5.4 Las unidades de eritrocitos que se utilicen para transfusión de pacientes con presencia de anticuerpos irregulares deberán carecer de los antígenos correspondientes a los anticuerpos identificados, para ello las unidades deberán ser estudiadas con antisueros de fuentes confiables por medio de técnicas validadas. Para la investigación de antígenos cuyo antisuero no se encuentra de manera comercial se podrá utilizar un suero de alguna seroteca. Se podrán utilizar técnicas de biología molecular para buscar unidades fenotipo deducido compatibles (deducido por genotipo) en pacientes con anticuerpos irregulares.
10.5.5.5 Cuando se encuentre una unidad de eritrocitos fenotipo compatible que será transfundida a un paciente con anticuerpos irregulares, deberá realizarse la prueba cruzada mayor con cada unidad a transfundir.
10.5.5.6 En pacientes con presencia de autoanticuerpos que causen interferencia en las pruebas será necesario realizar técnicas de separación de autoanticuerpos de los posibles aloanticuerpos. En estos pacientes, las pruebas cruzadas, el rastreo de anticuerpos irregulares y en su caso la identificación, deberán realizarse con suero del paciente sin autoanticuerpos.
10.5.5.7 Los pacientes y donadores con presencia de anticuerpos irregulares de importancia clínica, deberán ser informados por escrito. El resultado para los pacientes quedará en el expediente clínico y para los donadores en la historia clínica del donador.
10.5.5.8 En caso de pacientes regulares deberá compararse el resultado de rastreo e identificación de anticuerpos con los registros de visitas previas o con informes de resultados que el paciente se haya realizado en otros centros. Estos registros deberán tomarse en cuenta al momento de la transfusión ya que puede haber anticuerpos evanescentes que no sean detectados en la muestra del paciente.
10.5.6 Pruebas cruzadas
10.5.6.1 Las pruebas cruzadas deberán incluir:
10.5.6.1.1 Identificación o ratificación del grupo ABO y Rh (D) tanto en el donador como en el receptor.
10.5.6.1.2 Prueba mayor. Permite demostrar la presencia o ausencia de anticuerpos regulares o irregulares en el suero del receptor contra antígenos presentes en los eritrocitos del donante;
10.5.6.1.3 La investigación de anticuerpos irregulares de importancia clinica en donadores.
10.5.6.1.4 Autotestigo. En la realización de las pruebas mayor y menor.
10.5.6.2 Las pruebas cruzadas tendrán una vigencia máxima de 72 horas cuando:
10.5.6.2.1 En los últimos tres meses el receptor tenga antecedentes propiciadores de aloinmunización, tales como embarazo o transfusiones, o
10.5.6.2.2 Cuando el receptor hubiese recibido una transfusión después de la realización de la prueba cruzada. De existir cualquiera de estas situaciones la prueba cruzada deberá repetirse.
10.5.6.3 En las situaciones que se señalan a continuación, las pruebas cruzadas se efectuarán con las variaciones siguientes:
10.5.6.3.1 Cuando un receptor tuviese una prueba de antiglobulina directa positiva, las pruebas cruzadas se realizarán empleando el suero del receptor y un eluido de los eritrocitos del mismo, y
10.5.6.3.2 Cuando se transfunda plasma no se requiere hacer la prueba cruzada mayor siguiendo las indicaciones de la Tabla 25 de este proyecto de Norma.
10.5.6.3.3 No será necesario realizar prueba cruzada mayor, cuando se transfundan concentrados de eritrocitos si éstos son compatibles por medio de "cruce electrónico" o" prueba cruzada electrónica".
10.5.6.4 Los Bancos de Sangre y Servicios de Transfusión Hospitalario deberán contar con procedimientos normalizados de operación que especifiquen como deben actuar en casos de urgencias transfusionales.
10.5.6.5 La urgencia transfusional acreditada por el médico tratante y avalada por el médico responsable del Banco de Sangre o del Servicio de Transfusión Hospitalario, no exime la práctica de las pruebas cruzadas de compatibilidad; sin embargo, la sangre o concentrado de eritrocitos podrán liberarse anticipadamente para su transfusión, hasta haber corroborado el grupo ABO y Rh (D) de la unidad y de receptor y verificar la compatibilidad ABO, mediante una prueba rápida en medio salino.
10.5.6.6 Entre tanto se continuará con las pruebas de compatibilidad utilizando un medio facilitador de la reacción, preferentemente solución de baja fuerza iónica (solución de "liss") para abreviar el tiempo. Las pruebas cruzadas se llevarán hasta su término con la prueba de antiglobulina humana.
De detectarse incompatibilidad en las pruebas cruzadas, el Banco de Sangre o, en su caso, el Servicio de Transfusión Hospitalario, deberá dar aviso inmediatamente, con la finalidad de evitar o interrumpir la transfusión.
10.5.7 Las pruebas de hemocompatibilidad previas a una transfusión en neonatos y menores de cuatro meses de edad:
10.5.7.1 Técnica de elución: Se emplearán el suero o plasma de la madre. En caso de carecer de muestras maternas, se deberán efectuar con un eluido de los eritrocitos del menor.
10.5.7.2 La técnica empleada debe ser validada y adecuada para investigación de anticuerpos contra antígenos de importancia clínica.
10.5.7.3 En caso de incompatibilidad materno-fetal de tipo ABO, el eluido también deberá enfrentarse con células A y B.
Se deberá contar con un Procedimiento Normalizado de Operación que establezca las técnicas que se deben emplear para el manejo y resolución de estas pruebas.
10.5.8 Pruebas de compatibilidad electrónicas.
10.5.8.1 Para la utilización de las pruebas de compatibilidad electrónicas debe demostrarse la ausencia de anticuerpos irregulares en la muestra actual y en registros históricos.
10.5.8.2 Las pruebas de compatibilidad electrónicas deben cumplir los siguientes requisitos:
10.5.8.2.1 El grupo ABO de los concentrados de eritrocitos debe haberse comprobado en el banco de sangre o servicio de transfusión y los resultados deben coincidir con los datos en la etiqueta de la bolsa. Esta información debe ser trazable.
10.5.8.2.2 La identificación de la muestra pretransfusional del paciente debe realizarse por un método electrónico (código de barras o similar).
10.5.8.2.3 El grupo ABO y Rh (D) del receptor debe realizarse mediante una técnica automatizada.
10.5.8.2.4 La transferencia del resultado del grupo ABO y Rh (D) de la muestra pretransfusional desde el analizador a la historia del paciente en el sistema electrónico debe realizarse de forma electrónica.
10.5.8.2.5 El sistema debe verificar el resultado del grupo ABO de la muestra pretransfusional con los resultados previos del mismo paciente y debe avisar al operador en caso de discrepancia.
10.5.8.2.6 En el momento de la asignación informática del componente al receptor, el sistema debe verificar el grupo ABO de ambos y debe disponer de un sistema de alarma que avise en caso de discrepancia e impida la asignación cuando exista incompatibilidad.
10.5.8.2.7 El sistema de pruebas de compatibilidad electrónica debe estar validado.
10.5.8.2.8 El Servicio de Transfusión Hospitalario debe disponer de un procedimiento que evite la salida para transfusión de cualquier unidad de concentrado eritrocitario que no haya sido asignada al paciente en el sistema y que no haya pasado, por tanto, por la prueba cruzada electrónica.
11 Identificación de las unidades y de las muestras sanguíneas
11.1 El Banco de Sangre, Centro de Procesamiento de Sangre, Centro de Colecta, Centro de Distribución, Servicio de Transfusión Hospitalario y Centro de Calificación Biológica, deberán contar con un proceso que asegure el correcto etiquetado e identificación de todas las unidades de sangre, productos sanguíneos, mezclas de componentes y las muestras de sangre, plasma o suero.
11.2 Las etiquetas de los productos sanguíneos y de las muestras deben estar firmemente adheridas y ser fácilmente legibles.
Podrán emplearse etiquetas validadas con sistemas electrónicos que permitan ingresar y verificar los datos, procesamiento, temperatura y trazabilidad inherente a las unidades.
11.3 Para identificar las unidades, sus correspondientes muestras y todos los documentos y registros relativos a ellas, se empleará un sistema numérico o alfanumérico que permita la trazabilidad de cada unidad de sangre y de sus componentes; desde su extracción hasta su transfusión o, o en su caso, suministro para elaboración de hemoderivados o, o bien, su destino final.
11.4 La identificación numérica o alfanumérica será exclusiva y deberá permanecer inalterada para cada unidad, sus muestras, documentos y registros. Se debe utilizar el sistema de código de barras, código QR u otros de mejor tecnología, que admitan el enlace con otros sistemas electrónicos tales como: dispositivos electrónicos o de radiofrecuencia. A toda unidad proveniente de algún Servicio de Sangre, se le deberá agregar la identificación numérica o alfanumérica correspondiente al establecimiento al cual ingresa, permitiendo en todo momento la trazabilidad de la unidad o la muestra.
11.5 Todos los Servicios de Sangre deberán contar con procedimientos escritos para evitar los errores durante el etiquetado o identificación de las unidades, sus muestras correspondientes, documentos y registros. Cuando un componente sanguíneo se transfiera a otra bolsa, los procedimientos deberán asegurar la correcta adjudicación de la identificación de la unidad desde la bolsa original hasta la bolsa definitiva.
11.6 Los tubos que contienen las muestras de sangre, plasma o suero de donantes y de pacientes, para efectos de realización de las pruebas de detección de enfermedades transmisibles por transfusión, hemoclasificación, hemocompatibilidad u otras, deberán estar debidamente rotulados para su correcta identificación y su etiqueta contendrá anotada, como mínimo, la información siguiente:
11.6.1 Nombre del donante o del paciente;
11.6.2 Fecha y hora en que la muestra fue tomada;
11.6.3 En caso de donantes, el número exclusivo asignado a la unidad de sangre o productos sanguíneos colectados, y
11.6.4 Tratándose de pacientes o receptores, en su caso, nombre completo, fecha de nacimiento contemplando día, mes y año en el formato dd/mm/aaaa, número de expediente o registro, y el nombre del servicio clínico.
11.7 Unidades no procesadas.
Las etiquetas de las bolsas primarias de sangre total para fines de estudio y fraccionamiento en sus diversos componentes y las unidades de productos sanguíneos obtenidas por aféresis, deberán contener como mínimo la información siguiente:
11.7.1 Nombre del Banco de Sangre o Centro de Colecta donde la unidad fue colectada;
11.7.2 El nombre del componente sanguíneo;
11.7.3 Identificación numérica o alfanumérica exclusiva de la unidad;
11.7.4 Nombre de la solución anticoagulante o, o en su caso, el nombre y volumen de la solución aditiva;
11.7.5 Fecha de la extracción;
11.7.6 Hora de inicio y de término de la recolección;
11.7.7 Volumen de la unidad, y
11.8 El rango de temperatura en que deben conservarse y transportarse, se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C). Identificación de unidades procesadas y estudiadas:
11.8.1 Sangre y concentrados de eritrocitos:
11.8.1.1 Unidades de sangre y concentrados de eritrocitos. Las etiquetas de las unidades de sangre y concentrados de eritrocitos deberán contener la información siguiente:
11.8.1.1.1 Nombre del Servicio de Sangre procesador. Tratándose de unidades de sangre total que fuesen a emplear en transfusión, en su caso, podrá omitirse el nombre del centro de colecta donde la unidad se hubiese recolectado, mientras se cuente con los registros que permitan la trazabilidad;
11.8.1.1.2 Nombre del componente sanguíneo que se trate;
11.8.1.1.3 La identificación numérica o alfanumérica exclusiva de la unidad. Tratándose de concentrados de eritrocitos derivados de una doble colecta en una sola sesión de aféresis, además de la identificación exclusiva, será un número adicional que permita su identificación, por ejemplo: "aféresis 1", "aféresis 2";
11.8.1.1.4 Nombre de la solución anticoagulante, o en su caso, el nombre y volumen de la solución aditiva;
11.8.1.1.5 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.1.1.6 Grupo sanguíneo ABO y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas "POSITIVO" o "NEGATIVO" o con los símbolos o siglas "+" o "NEG", respectivamente;
11.8.1.1.7 Los resultados de fenotipos de grupo sanguíneo distintos a los del sistema ABO y del Rh (D), de haberse efectuado;
11.8.1.1.8 Tratándose de concentrado de eritrocitos y de haberse detectado anticuerpos irregulares, se indicará su especificidad;
11.8.1.1.9 Fechas y horas de extracción y de caducidad;
11.8.1.1.10 Volumen aproximado de la unidad;
11.8.1.1.11 El rango de temperatura en que deben conservarse y transportar, se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C);
11.8.1.1.12 Los señalamientos siguientes:
11.8.1.1.12.1 No transfundirse en presencia de hemólisis o cualquier otro signo de deterioro;
11.8.1.1.12.2 No mezclarse con medicamentos o soluciones que no sea solución salina isotónica al 0.9 %, y
11.8.1.1.12.3 Transfundirse a través de un filtro de 170 - 200 micras (µm) o en su caso, un filtro para leucodepletar, y
11.8.1.1.13 En su caso, contendrá la información siguiente:
11.8.1.1.13.1 Los procedimientos adicionales que se hubiesen efectuado, tales como irradiación, remoción de la capa leucoplaquetaria o filtrado para leucodepleción, y
11.8.1.1.13.2 Que las pruebas de compatibilidad no se han completado, cuando así se hubiese requerido ante una urgencia transfusional.
11.8.1.2 Unidades de sangre reconstituida:
Las etiquetas de la sangre reconstituida con eritrocitos y plasma de distintos donantes (mezcla de componentes) deberán contener la información siguiente:
11.8.1.2.1 Nombre del Servicio de Sangre procesador
11.8.1.2.2 Nombre del componente sanguíneo de que se trate;
11.8.1.2.3 La identificación numérica o alfanumérica de cada unidad integrante, o bien, una identificación numérica o alfanumérica exclusiva de la mezcla, en este último caso, los registros del Banco de Sangre, Centro de Procesamiento de Sangre o Servicio de Transfusión Hospitalario, procesador deberán, permitir fácil e inequívocamente la trazabilidad de cada una de las unidades que integran la mezcla;
11.8.1.2.4 Nombre de la solución anticoagulante o en su caso, el nombre y volumen de la solución aditiva;
11.8.1.2.5 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.1.2.6 El grupo sanguíneo ABO y Rh (D) de los eritrocitos y del plasma. La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas "POSITIVO" o "NEGATIVO", o con los símbolos o siglas "+" o "NEG", respectivamente;
11.8.1.2.7 Los resultados de fenotipos de grupo sanguíneo distintos a los del sistema ABO y del Rh (D), de haberse efectuado;
11.8.1.2.8 Fechas de extracción y de caducidad de componentes que integran la mezcla y, en su caso, la nueva fecha y hora de caducidad posterior a la reconstitución;
11.8.1.2.9 Volumen aproximado de la unidad;
11.8.1.2.10 El rango de temperatura en que deben conservarse y transportarse se señalará con números y signos por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C).
11.8.1.2.11 Los señalamientos siguientes:
11.8.1.2.11.1 No transfundirse en presencia de hemólisis o cualquier otro signo de deterioro;
11.8.1.2.11.2 No mezclarse con medicamentos o soluciones que no sea solución salina isotónica al 0.9%, y
11.8.1.2.11.3 Transfundirse a través de un filtro de 170 - 200 micras (µm) en su caso, un filtro para leucodepletar, y
11.8.1.2.12 En su caso, los procedimientos adicionales que se hubiesen efectuado, tales como irradiación, remoción de la capa leucoplaquetaria o leucodepleción.
11.8.1.3 Unidades de eritrocitos lavados:
Las etiquetas de las unidades de eritrocitos lavados, deberán contener la información siguiente:
11.8.1.3.1 Nombre del Banco de Sangre, Centro de Procesamiento de Sangre o Servicio de Transfusión Hospitalario procesador;
11.8.1.3.2 Nombre del componente sanguíneo que se trata: "eritrocitos lavados";
11.8.1.3.3 La identificación numérica o alfanumérica exclusiva de la unidad. Tratándose de concentrados de eritrocitos derivados de una doble recolección de una sesión de aféresis, además de la identificación única, un número adicional que permita su identificación, por ejemplo: "aféresis 1", "aféresis 2", etcétera;
11.8.1.3.4 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.1.3.5 Grupo sanguíneo ABO y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas: "POSITIVO" o "NEGATIVO", o con los símbolos o siglas "+" o "NEG", respectivamente;
11.8.1.3.6 Los resultados de fenotipos de grupo sanguíneo distintos a los del sistema ABO y del Rh (D), de haberse efectuado;
11.8.1.3.7 La fecha del lavado de la unidad y posterior a este, lo que se indica a continuación:
11.8.1.3.8 La nueva fecha de caducidad y la hora de la misma;
11.8.1.3.9 El nombre, volumen y concentración de la solución en la que se resuspendieron los eritrocitos, y
11.8.1.3.10 El hematocrito final de la unidad;
11.8.1.3.11 Volumen aproximado del producto final;
11.8.1.3.12 El rango de temperatura en que deben conservarse y transportarse se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C);
11.8.1.3.13 Los señalamientos siguientes:
11.8.1.3.13.1 No transfundirse en presencia de hemólisis o cualquier otro signo de deterioro;
11.8.1.3.13.2 No mezclarse con medicamentos o soluciones que no sea solución salina isotónica al 0.9%,
11.8.1.3.13.3 Los procedimientos adicionales que se hubiesen efectuado, tales como irradiación, remoción de la capa leucoplaquetaria o filtrado para leucodepleción.
11.8.1.3.13.4 Que las pruebas de compatibilidad no se han completado, cuando así se hubiese requerido ante una urgencia transfusional, y:
11.8.1.3.13.5 Transfundirse a través de un filtro de 170 - 200 micras (µm) en su caso, un filtro para leucodepletar.
11.8.1.4 Unidades de eritrocitos congeladas y unidades de eritrocitos descongeladas:
Las etiquetas de las unidades de eritrocitos congelados y de las unidades descongeladas y lavadas, deberán contener la información siguiente:
11.8.1.4.1 Nombre del Banco de Sangre, Centro de Procesamiento de Sangre o Servicio de Transfusión Hospitalario procesador;
11.8.1.4.2 Nombre del componente sanguíneo que se trate;
11.8.1.4.3 La identificación numérica o alfanumérica exclusiva de la unidad. Tratándose de concentrados de eritrocitos derivados de una doble recolección de una sesión de aféresis, además de la identificación única, un número adicional que permita su identificación, por ejemplo: "aféresis 1", "aféresis 2", etcétera;
11.8.1.4.4 Resultado de las pruebas de detección de los agentes infecciosos.
11.8.1.4.5 Grupo sanguíneo AB0 y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas: "POSITIVO" o "NEGATIVO", o con los símbolos o siglas "+" o "NEG", respectivamente;
11.8.1.4.6 Los resultados de fenotipos de grupo sanguíneo distintos a los del sistema AB0 y del Rh (D), de haberse efectuado;
11.8.1.4.7 De haberse detectado anticuerpos irregulares, se indicará su especificidad;
11.8.1.4.8 Nombre, volumen y concentración de la solución crioprotectora;
11.8.1.4.9 La fecha de preparación y congelación;
11.8.1.4.10 Volumen aproximado de la unidad;
11.8.1.4.11 El rango de temperatura en que deben conservarse y transportarse se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C);
11.8.1.4.12 Tras el descongelamiento y lavado de la unidad, se deberá modificar en la etiqueta:
11.8.1.4.12.1 Fecha y hora de caducidad;
11.8.1.4.12.2 La temperatura de conservación y transportación se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C);
11.8.1.4.12.3 El nombre, volumen y concentración de la solución en la que se resuspendieron los eritrocitos, y
11.8.1.4.12.4 El hematocrito final de la unidad, y
11.8.1.4.13 Los señalamientos siguientes:
11.8.1.4.13.1 No transfundirse en presencia de hemólisis o cualquier otro signo de deterioro;
11.8.1.4.13.2 No mezclarse con medicamentos o soluciones que no sea solución salina isotónica al 0.9%, y
11.8.1.4.13.3 Transfundirse a través de un filtro de 170 - 200 micras (µm).
11.8.2 Preparados con plaquetas:
11.8.2.1 Unidades de plaquetas recuperadas o mezcla de éstas.
Las etiquetas de las unidades de plaquetas recuperadas o mezclas de éstas deberán contener la información siguiente:
11.8.2.1.1 Nombre del Banco de Sangre, Centro de Procesamiento de Sangre o Servicio de Transfusión Hospitalario procesador;
11.8.2.1.2 Nombre del componente sanguíneo que se trate: "unidad de plaquetas recuperadas o mezcla de plaquetas";
11.8.2.1.3 La identificación numérica o alfanumérica exclusiva de la unidad de plaquetas recuperada. Tratándose de mezclas de plaquetas se deberá indicar:
11.8.2.1.3.1 La cantidad de unidades de plaquetas recuperadas que integran la mezcla, y
11.8.2.1.3.2 La identificación numérica o alfanumérica exclusiva asignada a la mezcla. Los registros del Banco de Sangre, Centro de Procesamiento de Sangre o Servicio de Transfusión Hospitalario, deberán permitir fácil e inequívocamente la trazabilidad de cada una de las unidades de plaquetas que integran la mezcla;
11.8.2.1.3.3 Nombre de la solución anticoagulante o, en su caso, el nombre y volumen de la solución aditiva;
11.8.2.1.3.4 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.2.1.3.5 Grupo sanguíneo ABO y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas: "POSITIVO" o "NEGATIVO" o con los símbolos o siglas "+" o "NEG", respectivamente;
11.8.2.1.3.6 De haberse detectado anticuerpos irregulares, se indicará su especificidad;
11.8.2.1.3.7 Fecha preparación y caducidad de la mezcla;
11.8.2.1.3.8 El contenido de plaquetas en la unidad o mezcla ya sea calculado o el conteo real;
11.8.2.1.3.9 El rango de temperatura y condiciones en que debe conservarse se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C);
11.8.2.1.4 Los señalamientos siguientes:
11.8.2.1.4.1 No mezclarse con medicamentos, y
11.8.2.1.4.2 Transfundirse a través de un filtro de 170 - 200 micras (µm), y
11.8.2.1.4.3 En su caso, los procedimientos adicionales que se hubiesen efectuado, tales como lavado, irradiación, leucodepleción o cualquier otro.
11.8.2.2 Unidades de plaquetas obtenidas por aféresis:
Las etiquetas de las unidades de plaquetas obtenidas por aféresis deberán contener la información siguiente:
11.8.2.2.1 Nombre del Banco de Sangre, procesador;
11.8.2.2.2 Nombre del componente sanguíneo que se trate: "unidad de plaquetas de aféresis";
11.8.2.2.3 La identificación numérica o alfanumérica exclusiva de la unidad. De haberse obtenido varias unidades de plaquetas en una sesión de aféresis, además de la identificación única, un número adicional que permita identificarlas, por ejemplo: "aféresis 1", aféresis 2", etcétera;
11.8.2.2.4 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.2.2.5 Grupo sanguíneo ABO y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas: "POSITIVO" o "NEGATIVO" o con los símbolos o siglas "+" o "NEG", respectivamente;
11.8.2.2.6 De haberse detectado anticuerpos irregulares, se indicará su especificidad;
11.8.2.2.7 Fechas de extracción y de caducidad;
11.8.2.2.8 Nombre de la solución anticoagulante, o en su caso, el nombre y volumen de la solución aditiva;
11.8.2.2.9 El contenido de plaquetas en la unidad ya sea calculado o el conteo real;
11.8.2.2.10 El rango de temperatura y condiciones en que debe conservarse se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C);
11.8.2.2.11 De haberse efectuado la genotipificación de antígenos leucocitarios y plaquetarios, deberá incluirse los antígenos detectados;
11.8.2.2.12 En su caso, los procedimientos adicionales que se hubiesen efectuado, tales como lavado, irradiación, leucodepleción, y
11.8.2.2.13 Los señalamientos siguientes:
11.8.2.2.13.1 No mezclarse con medicamentos, y
11.8.2.2.13.2 Transfundirse a través de un filtro de 170 - 200 micras (µm).
11.8.3 Unidades de granulocitos:
Las etiquetas de las unidades de granulocitos deberán contener la información siguiente:
11.8.3.1 Nombre Banco de Sangre procesador;
11.8.3.2 El nombre del componente sanguíneo (granulocitos de aféresis);
11.8.3.3 La identificación numérica o alfanumérica exclusiva de la unidad;
11.8.3.4 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.3.5 Grupo sanguíneo ABO y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas: "POSITIVO" o "NEGATIVO" o con los símbolos o siglas "+" o "NEG", respectivamente;
11.8.3.6 Fecha de la extracción;
11.8.3.7 Fecha y hora de caducidad;
11.8.3.8 Nombre de la solución anticoagulante, soluciones aditivas y en su caso de otros agentes;
11.8.3.9 El número de granulocitos contenido en la unidad;
11.8.3.10 El rango de temperatura en que debe conservarse, se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C);
11.8.3.11 De haberse efectuado el tipo HLA, y
11.8.3.12 Los señalamientos siguientes:
11.8.3.12.1 No mezclarse con medicamentos, y
11.8.3.12.2 Transfundirse a través de un filtro de 170-200 micras (µm).
11.8.4 Plasma y crioprecipitados:
11.8.4.1 Unidades de plasma.
Las etiquetas de las unidades de plasma deberán contener la información siguiente:
11.8.4.1.1 Nombre del Banco de Sangre o Centro de Procesamiento de Sangre procesador;
11.8.4.1.2 Nombre del componente sanguíneo, incluyendo si procede de sangre total o de aféresis;
11.8.4.1.3 La identificación numérica o alfanumérica exclusiva de la unidad. En caso de varias unidades de plasma obtenidas en una sesión de aféresis, además de la identificación exclusiva, un número adicional que permita identificarlas, por ejemplo: "aféresis 1", "aféresis 2", etcétera;
11.8.4.1.4 Nombre de la solución anticoagulante;
11.8.4.1.5 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.4.1.6 Grupo sanguíneo ABO y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas: "POSITIVO" o "NEGATIVO" o con sus los símbolos o siglas "+" o "NEG", respectivamente;
11.8.4.1.7 Fecha de la extracción;
11.8.4.1.8 Fecha de caducidad, modificándola después del descongelamiento y agregando la hora de caducidad;
11.8.4.1.9 El volumen de la unidad;
11.8.4.1.10 El rango de temperatura en que debe conservarse se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C), modificándola después del descongelamiento;
11.8.4.1.11 En su caso, el señalamiento de los procedimientos adicionales que se hubiesen efectuado, tales como:
11.8.4.1.11.1 Si está en periodo de cuarentena o después de éste;
11.8.4.1.11.2 Si fue sometido a leucodepleción previa al almacenamiento, y
11.8.4.1.12 Los señalamientos siguientes:
11.8.4.1.12.1 No mezclarse con medicamentos, y
11.8.4.1.12.2 Transfundirse a través de un filtro de 170-200 micras (µm).
En caso de que las unidades de plasma se egresen congeladas, se acompañarán con las instrucciones para su descongelamiento.
11.8.4.2 Unidades o mezclas de crioprecipitados.
Las etiquetas de las unidades o mezclas de crioprecipitados deberán contener la información siguiente:
11.8.4.2.1 Nombre del Banco de Sangre, Centro de Procesamiento de Sangre o Servicio de Transfusión Hospitalario procesador;
11.8.4.2.2 Nombre del componente sanguíneo (crioprecipitado de plasma fraccionado o de aféresis o bien, mezcla de crioprecipitados);
11.8.4.2.3 La identificación numérica o alfanumérica exclusiva de la unidad. En caso de varias unidades de crioprecipitados obtenidas en una sesión de plasmaféresis, además de la identificación única, un número adicional que permita identificarlas, por ejemplo: "aféresis 1", "aféresis 2", etcétera;
11.8.4.2.4 Tratándose de una mezcla de crioprecipitados, la cantidad de unidades que la integran y la identificación numérica o alfanumérica exclusiva de la mezcla. Los registros del Banco de Sangre deberán permitir fácil e inequívocamente la trazabilidad de cada una de las unidades que integran la mezcla;
11.8.4.2.5 Nombre de la solución anticoagulante;
11.8.4.2.6 Resultado de las pruebas de detección de los agentes infecciosos;
11.8.4.2.7 Grupo sanguíneo ABO;
11.8.4.2.8 Fecha de la preparación;
11.8.4.2.9 Fecha y hora de caducidad, modificándola después del descongelamiento y agregando la hora de caducidad;
11.8.4.2.10 El volumen aproximado de la unidad o mezcla;
11.8.4.2.11 Temperatura de conservación, se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C; + 20°C a + 24°C; - 20°C a - 25°C), modificándola después del descongelamiento;
11.8.4.2.12 En su caso, el señalamiento de los procedimientos adicionales que se hubiesen efectuado, tales como:
11.8.4.2.12.1 Si está en periodo de cuarentena o después de éste;
11.8.4.2.12.2 Si fue sometido a leucodepleción previa al almacenamiento, y
11.8.4.2.13 Los señalamientos siguientes:
11.8.4.2.13.1 No mezclarse con medicamentos, y
11.8.4.2.13.2 Transfundirse a través de un filtro de 170-200 micras (µm).
En caso de que las unidades o mezclas de crioprecipitados se egresen en estado de congelación, se acompañarán con las instrucciones para su descongelamiento.
11.9 Productos sanguíneos irradiados.
La etiqueta de los productos sanguíneos irradiados deberá indicar, además de la información concerniente al producto sanguíneo de que se trate, lo siguiente:
11.9.1 Que el producto sanguíneo fue irradiado, la fecha y la dosis de radiación aplicada;
11.9.2 El tipo de fuente de irradiación empleado;
11.9.3 Tratándose de concentrados de eritrocitos, en su caso, la nueva fecha de caducidad, de conformidad a lo dispuesto en el inciso 9.8.10 de este proyecto de Norma, y
11.9.4 Tratándose para transfusión intrauterina o para un neonato, el señalamiento que deberá transfundirse en el lapso de las 24 horas que siguen a la irradiación.
11.10 De no haber espacio suficiente en las etiquetas de las unidades para el registro de la información que solicita este capítulo, ésta deberá incluirse en un marbete anexo a la unidad.
11.11 Para la identificación de las unidades de sangre y productos sanguíneos para uso autólogo, véase el inciso 13.2.16 de este proyecto de Norma.
11.12 A las unidades o mezclas de productos sanguíneos que hubiesen concluido su periodo de vigencia o cualquier otra eventualidad que motive su destino final, se les anotará en su etiqueta la leyenda: "BAJA", "NO TRANSFUNDIRSE" o cualquier otra medida que garantice su exclusión del uso terapéutico, en tanto se le da destino final, a la brevedad.
12 Selección de unidades de sangre y productos sanguíneos para uso transfusional
12.1 Las unidades de sangre y productos sanguíneos deberán transfundirse preferentemente a receptores de grupo ABO idéntico (isogrupo).
12.2 Para la transfusión de preparados que contengan eritrocitos, se deberán observar las disposiciones siguientes:
12.2.1 Las señaladas en las Tablas 25 y 26 de este proyecto de Norma.
Tabla 25
Disposiciones para la transfusión de concentrados de eritrocitos y sangre reconstituida
Unidades o mezclas |
Opciones, en orden de preferencia para la aplicación de unidades, en lo relativo al sistema ABO |
Pruebas cruzadas |
Concentrado de eritrocitos |
a) Isogrupo, o |
La prueba mayor deberá ser compatible |
b) No isogrupo, siempre que los eritrocitos de la unidad sean compatibles en el sistema ABO con el plasma del receptor y conforme al orden de preferencia que señala la tabla 30 de este proyecto de Norma. |
||
Sangre reconstituida |
Los eritrocitos y el plasma de la unidad serán compatibles entre ellos y con el receptor. |
Tabla 26
Opciones, en orden de preferencia, para la transfusión de concentrado de eritrocitos compatibles en los sistemas AB0 y Rh (D) en adultos y niños mayores de 4 meses.
Grupo del paciente/receptor |
Primera |
Segunda |
Tercera |
Cuarta |
O positivo |
O positivo |
O negativo |
Inexistente |
Inexistente |
O negativo |
O negativo |
Inexistente |
Inexistente |
Inexistente |
A positivo |
A positivo |
A negativo |
O positivo |
O negativo |
A negativo |
A negativo |
0 negativo |
Inexistente |
Inexistente |
B positivo |
B positivo |
B negativo |
O positivo |
O negativo |
B negativo |
B negativo |
0 negativo |
Inexistente |
Inexistente |
AB positivo |
AB positivo o negativo |
A positivo o negativo |
B positivo o negativo |
O positivo o negativo |
AB negativo |
AB negativo |
A negativo |
B negativo |
O negativo |
Nota: Para la transfusión de preparados de eritrocitos Rh (D) positivos a receptores Rh (D) negativos véase el inciso 12.2.3 de este proyecto de Norma.
12.2.2 Al transfundir eritrocitos de grupo A, B o O a un receptor de grupo AB, se utilizará preferentemente eritrocitos de un solo grupo. De ser inevitable usar más de un grupo, los concentrados de eritrocitos deberán lavarse suficientemente, a menos que su contenido plasmático sea bajo, como en el caso de concentrado de eritrocitos con solución aditiva.
12.2.3 Los receptores Rh (D) positivos podrán recibir preparados de eritrocitos Rh (D) positivos o negativos. Los receptores Rh (D) negativos deberán recibir preparados de eritrocitos D negativos; sin embargo, en una urgencia transfusional y ante la carencia de eritrocitos D negativos, podrán transfundirse concentrados de eritrocitos Rh (D) positivos, siempre y cuando, se reúnan las condiciones siguientes:
12.2.3.1 Que los receptores no estuviesen previamente sensibilizados contra el antígeno D del sistema Rh y la prueba cruzada mayor resulte compatible, y
12.2.3.2 Que se cuente con la aprobación del responsable sanitario del Banco de Sangre o del servicio de transfusión y del médico tratante.
A los receptores Rh (D) negativos que se transfundan con preparados de eritrocitos Rh (D) positivos deberán recibir inmunoglobulina anti D en dosis suficientes para evitar la aloinmunización.
12.2.4 En pacientes de grupo A, B o AB que hubieran recibido transfusión masiva en la que se hubiesen empleado, entre otros, sangre total o plasma no isogrupo, se deberá investigar en su suero la presencia de anti A y anti B y de requerirse a corto plazo otras transfusiones, se utilizarán eritrocitos compatibles con el grupo ABO del plasma transfundido.
12.2.5 Los pacientes que recientemente hubiesen recibido múltiples transfusiones de concentrados de eritrocitos y en cualquier otro caso en que hubiese dos poblaciones de eritrocitos, en quienes la hemoclasificación se vea dificultada por la presencia de reacciones de campo mixto, los receptores deberán transfundirse con unidades de grupo O, o bien, transfundirse con unidades en las que se haya demostrado compatibilidad mediante la genotipificación efectuada en una fuente diferente a la sangre, por ejemplo: la saliva.
12.2.6 Cuando un receptor tenga anticuerpos irregulares de importancia clínica o antecedente de la presencia de tales anticuerpos, los preparados de eritrocitos a transfundir deberán ser compatibles y carecer de los antígenos correspondientes, excepto en circunstancias clínicas razonablemente justificadas y aprobadas por el médico responsable del Servicio de Sangre.
12.3 Para la transfusión de unidades de concentrados de plaquetas, de granulocitos, plasmas y crioprecipitados, así como de las mezclas de plaquetas y crioprecipitados, se observarán las disposiciones siguientes:
12.3.1 Los concentrados de plaquetas o mezcla de estas unidades se transfundirán conforme a lo siguiente:
12.3.1.1 Se emplearán unidades del mismo grupo ABO del receptor, salvo en las circunstancias que señala el inciso siguiente;
12.3.1.2 Cuando las existencias no permitan cubrir el requerimiento de plaquetas isogrupo, cualquier grupo ABO es aceptable. En estos casos es recomendable que las unidades o mezclas de plaquetas tengan bajo contenido plasmático o que estén suspendidas en solución salina isotónica al 0.9%, y
12.3.1.3 En caso de refractariedad a las plaquetas, se seleccionarán donantes cuyas plaquetas carezcan del o los antígenos contra los que reaccionan los anticuerpos presentes en el receptor.
12.3.2 No es recomendable transfundir plaquetas provenientes de donantes Rh (D) positivos a mujeres en edad reproductiva o menores de edad Rh (D) negativos, de ser necesario, deberá prevenirse la aloinmunización contra el antígeno D mediante la aplicación de globulina inmune anti D.
12.3.3 Para la transfusión de concentrados de granulocitos, se deberán emplear unidades del mismo grupo ABO del receptor.
12.3.4 Las unidades de concentrado de plaquetas o de granulocitos que tengan contaminación eritrocitaria macroscópicamente detectable (≥ 2 mL de eritrocitos por unidad), se transfundirán a receptores del mismo grupo ABO, o bien, las unidades serán compatibles con el plasma del receptor. En estos casos, se deberá demostrar, además, compatibilidad con la prueba cruzada mayor.
12.3.5 Los plasmas se transfundirán conforme a lo siguiente:
12.3.5.1 Preferentemente se emplearán unidades del mismo grupo ABO del receptor, y
12.3.5.2 De no haber en existencia unidades de plasma isogrupo, podrán emplearse unidades de distinto grupo ABO, siempre que éstas sean compatibles con los eritrocitos del receptor (véase la Tabla 27 de este proyecto de Norma).
Tabla 27
Opciones para la transfusión de plasma compatible en el sistema ABO y Rh
Grupo ABO y Rh (D) del receptor |
Orden de preferencia en cuanto al grupo ABO y Rh del plasma a transfundir |
|||
Primera |
Segunda |
Tercera |
Cuarta |
|
O positivo |
O positivo o negativo |
A positivo o negativo |
B positivo o negativo |
AB positivo o negativo |
O negativo |
O negativo o positivo |
A negativo o positivo |
B negativo o positivo |
AB negativo o positivo |
A positivo |
A positivo |
A negativo |
AB positivo |
AB negativo |
A negativo |
A negativo |
A positivo |
AB negativo |
AB positivo |
B positivo |
B positivo |
B negativo |
AB positivo |
AB negativo |
B negativo |
B negativo |
B positivo |
AB negativo |
AB positivo |
AB positivo |
AB positivo |
AB negativo |
Inexistente |
Inexistente |
AB negativo |
AB negativo |
AB positivo |
Inexistente |
Inexistente |
Nota: - Los plasmas de grupo Rh (D) negativos que se pretendan transfundir a receptores Rh (D) positivos deberán carecer de anticuerpos contra el antígeno D, conforme a lo establecido en el inciso 10.5.1.2 de este este Proyecto de Norma, y
- Los plasmas de grupo Rh (D) positivos que se pretendan transfundir a receptores Rh (D) negativos deberán carecer de contaminación eritrocitaria.
12.3.6 Para la transfusión de unidades de crioprecipitados o mezclas de éstas, cualquier grupo ABO es aceptable, sin embargo, es aconsejable que las unidades o mezclas de crioprecipitados no isogrupo tengan bajo contenido plasmático o que se reconstituyan con solución salina isotónica al 0.9 %.
12.3.7 Cuando los receptores tengan anticuerpos contra proteínas plasmáticas, incluyendo aquéllos con deficiencia de inmunoglobulina tipo A y con anticuerpos contra esta inmunoglobulina, se deberán transfundir unidades de concentrados de eritrocitos o de concentrados o mezclas de plaquetas lavadas.
12.3.8 Los receptores con antecedentes de exposiciones alogénicas múltiples tales como los politransfundidos o las mujeres con antecedentes de más de un embarazo deberán recibir componentes celulares leucodepletados.
12.4 Los receptores que se encuentren en los casos siguientes deberán recibir componentes celulares irradiados:
12.4.1 Fetos receptores de transfusiones intrauterinas;
12.4.2 Exsanguineotransfusión en prematuros y en recién nacidos de peso corporal inferior a 1,200 g;
12.4.3 Pacientes seleccionados inmunodeprimidos;
12.4.4 Inmunodeficiencia de células T;
12.4.5 Candidatos o receptores de trasplante de regeneración de médula ósea y hasta los seis meses tras haber efectuado un trasplante exitoso;
12.4.6 Receptores de unidades provenientes de familiares consanguíneos de primer y segundo grado;
12.4.7 Pacientes seleccionados con enfermedad de Hodgkin;
12.4.8 Cuando el receptor fuera a recibir transfusión de componentes HLA compatibles, y
12.4.9 Los demás que señala la guía de uso clínico de la sangre referenciada en el inciso 22.10 de este proyecto de Norma.
12.5 Los pacientes inmunodeprimidos y los neonatos de bajo peso deberán recibir productos sanguíneos de bajo riesgo para de citomegalovirus, toxoplasma y HTLV tipos I y II. Se consideran componentes de bajo riesgo los siguientes:
12.5.1 Componentes celulares obtenidos de donantes seronegativos a éstos agentes infecciosos;
12.5.2 Componentes celulares leucodepletados (mediante filtrado) hasta ≤1 x 106 leucocitos remantes por unidad;
12.5.3 Concentrado de eritrocitos desglicerolados, y
12.5.4 Componentes acelulares, tales como el plasma y el crioprecipitado.
12.6 Transfusión de urgencia. Para las transfusiones de urgencia se observarán las disposiciones siguientes:
Ante el desconocimiento del grupo ABO del paciente, se deberán transfundir concentrados de eritrocitos de grupo 0, preferentemente de grupo 0 negativo, siempre y cuando haya en existencia;
12.6.1 Los receptores con grupo ABO identificado, podrán recibir unidades isogrupo o bien de un grupo ABO compatible;
12.6.2 Las pruebas de compatibilidad deberán realizarse y completarse a la brevedad, aun cuando ya se hubiese iniciado la transfusión. En la bolsa del producto sanguíneo se deberá hacer constar que las pruebas de compatibilidad no se han completado, y
12.6.3 De carecer de concentrados de eritrocitos de grupo Rh (D) negativos, los receptores de este grupo podrán transfundirse con concentrados de eritrocitos Rh (D) positivos, siempre y cuando se observen las disposiciones que señala el inciso 12.2.3 de este proyecto de Norma.
12.7 Transfusión en neonatos y en receptores menores de cuatro meses de edad
12.7.1 Para la transfusión en neonatos y de receptores menores de cuatro meses de edad, se deberá observar lo siguiente:
12.7.2 Los neonatos y los menores de cuatro meses tienen anticuerpos circulantes correspondientes al sistema AB0 materno, lo que influye en la decisión del grupo sanguíneo del concentrado de eritrocitos que se pretende transfundir (véase la Tabla 28 de este proyecto de Norma).
Tabla 28
Opciones, en orden de preferencia, para la transfusión de concentrado de eritrocitos compatibles en los sistemas ABO y Rh (D) en recién nacidos y en menores de 4 meses
Grupo sanguíneo de la madre |
Grupo sanguíneo del menor |
Concentrado de eritrocitos que pueden transfundirse |
O negativo |
O positivo |
O negativo |
A negativo |
A positivo |
A negativo u O negativo |
B negativo |
B positivo |
B negativo u O negativo |
AB negativo |
AB positivo |
AB negativo, A negativo B, negativo u 0 negativo |
O positivo |
A positivo |
O positivo |
O positivo |
B positivo |
O positivo |
O positivo |
AB cis positivo |
O positivo |
O positivo |
A negativo |
O negativo |
O positivo |
B negativo |
O negativo |
O positivo |
AB cis negativo |
O negativo |
O positivo |
O negativo |
O negativo |
O positivo |
O positivo |
O positivo |
A positivo |
A positivo |
A positivo u O positivo |
B positivo |
B positivo |
B positivo u 0 positivo |
AB positivo |
AB positivo |
AB positivo, A positivo, B positivo u 0 positivo |
Nota: - AB cis significa que el grupo sanguíneo AB es codificado por un alelo del padre;
- De desconocerse el grupo sanguíneo materno se transfundirá concentrado de eritrocitos de grupo O y de Rh (D) idéntico al del menor.
12.7.2.1 Si el componente sanguíneo que se pretende transfundir procede de un donante consanguíneo en primer grado, se utilizarán componentes celulares leucodepletados e irradiados.
12.7.2.2 En neonatos con insuficiencia renal o hiperkalemia los productos sanguíneos irradiados se transfundirán antes de transcurridas 24 horas tras la irradiación.
12.7.2.3 En un receptor recién nacido o menor de cuatro meses, se procurará evitar la exposición alogénica a múltiples donantes, para ello es recomendable dividir el componente sanguíneo en volúmenes menores, (por ejemplo, entre 25 y 100 mL), empleando varias bolsas satélites y métodos que mantengan el sistema cerrado y por tanto la esterilidad del contenido.
12.7.2.4 Cuando fuesen a transfundirse productos sanguíneos cuyo fraccionamiento se hubiese realizado en sistema abierto, se deberán transfundir antes de 24 horas a partir del fraccionamiento si se hubieran mantenido entre +2º C y +6º C o antes de 6 horas si hubiesen conservado entre +20º y 24º C.
12.7.2.5 Cuando un receptor menor de cuatro meses vaya a transfundirse con cualquier unidad con eritrocitos de grupo distinto al O, deberá investigarse la presencia de anticuerpos anti-A y anti B en su suero o plasma y se transfundirán las unidades cuyos eritrocitos carezcan del antígeno contra el cual va dirigido el anticuerpo.
12.7.2.6 En presencia de anticuerpos irregulares de importancia clínica de origen materno, se deberán transfundir unidades con eritrocitos carentes del antígeno correspondiente o ser compatibles en la prueba de antiglobulina indirecta (Coombs indirecto).
De obtenerse un resultado positivo en la prueba de antiglobulina directa en la muestra sanguínea del menor, deberán transfundirse eritrocitos carentes del antígeno correspondiente a la especificidad del anticuerpo.
12.8 Exsanguineotransfusión. En enfermedad hemolítica del recién nacido (o enfermedad hemolítica perinatal) que requiera exsanguineotransfusión, se deberá proceder como sigue:
12.8.1 Cuando la enfermedad es por incompatibilidad ABO se deberán utilizar eritrocitos de grupo O con plasma del mismo grupo ABO del neonato o con plasma de grupo AB. Si es por incompatibilidad por grupo Rh (D), se deberá utilizar eritrocitos D negativos (véase la Tabla 29 de este proyecto de Norma).
Tabla 29
Elección de componentes para exsanguineotransfusión en enfermedad hemolítica del recién nacido por sensibilización materna en los sistemas ABO y Rh
Madre |
Neonato |
Grupo AB0 y Rh del concentrado de eritrocitos a transfundir |
Grupo AB0 y Rh del plasma fresco descongelado a transfundir |
O negativo |
O positivo |
O negativo |
O positivo o negativo |
A negativo |
A positivo |
A o O negativo |
A positivo o negativo o AB positivo o negativo |
B negativo |
B positivo |
B o O negativo |
B positivo o negativo o AB positivo o negativo |
AB negativo |
AB positivo |
AB, A, B o O negativo |
AB positivo o negativo |
O positivo |
A positivo |
O positivo |
A positivo, A negativo, AB positivo o AB negativo |
O positivo |
B positivo |
O positivo |
B positivo, B negativo, AB positivo o AB negativo |
O positivo |
AB cis positivo |
O positivo |
AB positivo o negativo |
B positivo |
A positivo |
O positivo |
AB positivo o negativo |
A positivo |
B positivo |
O positivo |
AB positivo o negativo |
Nota: Los antígenos del sistema Rh (D) no se encuentran en el plasma por lo que no requiere compatibilidad a este grupo.
12.8.2 Tratándose de incompatibilidad debida a otros sistemas antigénicos, se deberán utilizar eritrocitos carentes del antígeno responsable de la aloinmunización materna.
12.8.3 Se utilizará sangre reconstituida con plasma fresco descongelado hasta obtener un hematocrito final en la unidad o mezcla de entre 50% y 60%.
12.8.4 Los eritrocitos tendrán menos de 5 días de haberse extraído y preferentemente se habrán irradiado. Tratándose de prematuros y recién nacidos de peso corporal igual o inferior a 1,200 g, los eritrocitos habrán sido irradiados de conformidad con lo establecido en el inciso 9.8 de este proyecto de Norma.
12.8.5 Los eritrocitos irradiados deberán transfundirse antes de transcurridas las 24 horas que siguen a la irradiación.
12.9 Transfusión intrauterina
12.9.1 Transfusión intrauterina de concentrados de eritrocitos. La unidad deberá reunir los requisitos siguientes:
12.9.1.1 Los concentrados de eritrocitos no deberán tener más de cinco días de haberse extraído;
12.9.1.2 El hematocrito del concentrado de eritrocitos estará entre el 70% y 75%;
12.9.1.3 Los eritrocitos deberán ser de grupo O, Rh (D) negativos;
12.9.1.4 Los eritrocitos para transfundir carecerán del o los antígenos que reaccionen contra anticuerpos irregulares que la madre tuviese y que hubieran atravesado la barrera transplacentaria;
12.9.1.5 El componente será negativo en la investigación de citomegalovirus o estará leucodepletado mediante filtrado, y
12.9.1.6 El componente deberá estar irradiado y transfundido antes de transcurridas 24 horas tras la irradiación.
12.9.2 Los preparados de plaquetas para la transfusión intrauterina deberán reunir los requisitos siguientes:
12.9.2.1 Las plaquetas deberán concentrarse mediante la eliminación de parte del sobrenadante, se dejarán en reposo durante una hora y deberán transfundirse antes de transcurridas las 6 horas que siguen al procedimiento de concentración;
12.9.2.2 El componente será negativo en la investigación de citomegalovirus o estará leucodepletado mediante filtrado, y
12.9.2.3 El componente habrá sido irradiado.
12.9.3 Los neonatos que recibieron transfusión intrauterina que requieran transfusiones adicionales, deberán continuar recibiendo componentes celulares leucodepletados e irradiados.
12.10 Prevención de la inmunización al antígeno Rh (D).
12.10.1.1 El médico tratante será responsable de la indicación y la dosificación de la globulina inmune anti- D a fin de prevenir la aloinmunización al antígeno D.
12.11 Transfusión sanguínea en trasplante de regeneración de médula ósea.
12.11.1 Al receptor del trasplante se le deberá realizar, además del grupo sanguíneo ABO, Rh (D) y el escrutinio de anticuerpos irregulares contra eritrocitos, la prueba de antiglobulina humana directa (Coombs directo), estudios de linfocitotoxicidad y anticuerpos contra plaquetas en caso de que hubiere sospecha de refractariedad a éstas. En su caso, se hará comparación con estudios previos.
12.11.2 Para la aplicación de componentes celulares en la etapa previa al trasplante, se observará lo siguiente:
12.11.2.1 Las unidades de componentes celulares que se pretendan transfundir, no deberán provenir de familiares de primer o segundo grado;
12.11.2.2 A partir de los 30 días que anteceden al trasplante, se aplicarán componentes celulares irradiados, y
12.11.2.3 Los componentes que se vayan a transfundir estarán leucodepletados mediante filtrado.
12.11.3 Durante la etapa postrasplante de regeneración de medula ósea, los productos sanguíneos que se vayan a transfundir estarán irradiados y leucodepletados mediante filtrado.
12.11.4 Un receptor podrá recibir trasplante de células progenitoras de diferente grupo sanguíneo ABO. La incompatibilidad de grupo ABO podrá ser mayor, menor o mixta, conforme a lo señalado en la Tabla 30 de este proyecto de Norma:
Tabla 30
Incompatibilidad en el sistema ABO en trasplante de células progenitoras
Incompatibilidad mayor |
Incompatibilidad menor |
Incompatibilidad mixta (mayor y menor) |
|||
Grupo del receptor |
Grupo del donante de las CPr |
Grupo del receptor |
Grupo del donante de las CPr |
Grupo del receptor |
Grupo del donante de las CPr |
O |
A, B o AB |
A, B, o AB |
O |
A, B, o AB |
O |
A |
B o AB |
A o AB |
B |
A |
B |
B |
A o AB |
B o AB |
A |
B |
A |
12.11.5 En los receptores que vayan a recibir un trasplante de células progenitoras con incompatibilidad mayor, se deberá proceder como se indica a continuación:
12.11.5.1 Reducir al mínimo los eritrocitos presentes en la unidad de células progenitoras que se va a trasplantar, y
12.11.5.2 Previamente al trasplante, cuando el receptor tenga títulos de isohemaglutininas (IgM e IgG) mayores de 1:256, es recomendable reducir el título a 1:16 o menor, mediante plasmaféresis o cualquier otro método validado.
12.11.6 Ante requerimientos transfusionales en un paciente trasplantado con células progenitoras en las que exista incompatibilidad mayor, se deberán aplicar los productos sanguíneos que señala la Tabla 31 de este proyecto de Norma.
Tabla 31
Transfusión de productos sanguíneos en un paciente trasplantado con células progenitoras con incompatibilidad mayor en el sistema ABO
Grupo del receptor de CPr |
Grupo del donante de CPr |
Grupo AB0 de los concentrados de eritrocitos a transfundir |
Grupo AB0 del plasma o plaquetas a transfundir |
O |
A |
O |
A o AB |
O |
B |
O |
B o AB |
O |
AB |
O |
AB |
A |
AB |
A |
AB |
B |
AB |
B |
AB |
12.11.7 Los receptores de trasplante con incompatibilidad menor en los que donante de las células tenga un título de isohemaglutininas de ≥1:128, se reducirá al mínimo el plasma presente en la unidad de células progenitoras.
12.11.8 Ante requerimientos transfusionales en un paciente trasplantado con células progenitoras en las que exista incompatibilidad menor, se deberán aplicar los productos sanguíneos de conformidad con lo señalado la Tabla 32 de este proyecto de Norma.
Tabla 32
Transfusión de productos sanguíneos en un paciente trasplantado con células progenitoras con incompatibilidad menor en el sistema ABO
Grupo del receptor de CPr |
Grupo del donante de CPr |
Grupo AB0 de los concentrados de eritrocitos a transfundir |
Grupo AB0 del plasma o plaquetas a transfundir |
A |
O |
O |
A o AB |
B |
O |
O |
B o AB |
AB |
O |
O |
AB |
AB |
A |
A |
AB |
AB |
B |
B |
AB |
12.11.9 En los receptores de trasplante con incompatibilidad mixta se deberá proceder como sigue:
12.11.9.1 Se reducirá al mínimo el plasma y los eritrocitos de la unidad de células progenitoras, y
12.11.9.2 Previamente al trasplante, cuando el receptor tenga títulos de isohemaglutininas (IgM e IgG) mayores de 1:256, es recomendable reducir el título a 1:16 o menor mediante plasmaféresis o cualquier otro método validado.
12.11.10 Ante requerimientos transfusionales en un paciente trasplantado con células progenitoras en las que exista incompatibilidad mixta (mayor y menor), se deberán aplicar los productos sanguíneos que señala la Tabla 33 de este proyecto de Norma.
Tabla 33
Transfusión de productos sanguíneos en un paciente trasplantado con células progenitoras con incompatibilidad mixta (mayor y menor) en el sistema ABO
Grupo del receptor de CPr |
Grupo del donante de CPr |
Grupo AB0 de los concentrados de eritrocitos a transfundir |
Grupo AB0 del plasma o plaquetas a transfundir |
A |
B |
O |
AB |
B |
A |
O |
AB |
12.11.11 Después de un trasplante con incompatibilidad mayor o mixta, se continuarán las transfusiones de concentrados de eritrocitos de grupo 0 que se requieran hasta que el receptor cambie al grupo sanguíneo del donante de células progenitoras.
Asimismo, si después del trasplante el título de isohemaglutininas es > 1:16, se hará recambio plasmático con plasma de grupo AB o con soluciones coloides.
12.12 La transfusión de sangre total deberá evitarse.
13 Disposición de sangre y productos sanguíneos para uso autólogo
13.1 Disposiciones comunes:
13.1.1 La disposición de sangre y sus componentes para uso autólogo se podrá realizar mediante los procedimientos siguientes:
13.1.1.1 Depósito previo;
13.1.1.2 Procedimientos de reposición inmediata, que son:
13.1.1.2.1 Hemodilución aguda preoperatoria, y
13.1.1.2.2 Recuperación sanguínea transoperatoria, postoperatoria o ambas.
Los procedimientos de depósito previo y hemodilución aguda preoperatoria se podrán llevar a cabo mediante aféresis automatizada.
13.1.2 La indicación para la ejecución de un procedimiento de disposición de sangre para uso autólogo será responsabilidad del médico tratante.
El responsable sanitario de los Servicios de Sangre deberá fomentar la práctica de procedimientos de transfusión autóloga.
13.1.3 Para efectuar cualquier procedimiento de disposición de sangre para uso autólogo se deberá obtener la carta consentimiento informado del paciente o, en caso de menores o en estado de interdicción, del padre, la madre, el tutor, quien ejerza la patria potestad o el representante legal.
Los responsables de recabar la carta de consentimiento se señalan en el inciso 6.3.4 de este proyecto de Norma.
13.1.4 Las unidades recolectadas para uso autólogo solo podrán ser transfundidas al mismo donante. No deberán emplearse para uso alogénico ni destinarse para fraccionamiento para obtener hemoderivados.
13.2 Transfusión autóloga mediante procedimientos de depósito previo.
13.2.1 La transfusión autóloga mediante procedimientos de depósito previo se podrá efectuar en unidades médicas, que cuenten con Bancos de Sangre o Servicio de Transfusión Hospitalario, con personal capacitado y experimentado en la materia y con los equipos e insumos necesarios para efectuar las extracciones sanguíneas y en atender las reacciones adversas a la donación que los pacientes tuviesen.
13.2.2 En situaciones de cirugía programada cuando es previsible el requerimiento transfusional, el médico tratante que solicite un procedimiento de transfusión autóloga por depósito previo deberá indicar a los Servicios de Sangre lo siguiente:
13.2.2.1 Diagnóstico y tipo de intervención que se planea efectuar;
13.2.2.2 Número de unidades requeridas;
13.2.2.3 Fecha de la intervención, y
13.2.2.4 En su caso, los datos del establecimiento para la atención médica donde se efectuará la cirugía.
13.2.3 La disposición de sangre y productos sanguíneos mediante procedimientos de depósito previo será responsabilidad de los Servicios de Sangre, con excepción de la indicación del procedimiento y la supervisión de la transfusión, que serán responsabilidad del médico tratante o del médico que indique el procedimiento.
13.2.4 El personal médico calificado de los Servicios de Sangre o el responsable sanitario de los mismos será quien coordine todo el procedimiento y llevará a cabo, obligatoriamente, las actividades siguientes:
13.2.4.1 Proporcionar al donante la información concerniente al procedimiento de transfusión autóloga mediante depósito previo, obtener la firma de la carta de consentimiento informado y observar las demás disposiciones aplicables que señala el capítulo 6 denominado "Información, consentimientos y atención para donantes y receptores" de este proyecto de Norma;
13.2.4.2 Evaluar la aptitud del paciente para tolerar el procedimiento, de conformidad con los requisitos que para el efecto establece este capítulo, en coordinación con el médico tratante y, de ser necesario, se auxiliará de otros especialistas interconsultantes, de quienes deberá obtener su opinión escrita. La evaluación se llevará a cabo en privado y será de carácter confidencial;
13.2.4.3 Consignar en la historia clínica las actividades realizadas y los resultados obtenidos;
13.2.4.4 Supervisar la extracción, identificación, análisis, fraccionamiento, custodia y conservación de las unidades de sangre y de sus componentes;
13.2.4.5 Establecer para cada caso, en coordinación con el médico tratante, un programa de extracciones de acuerdo al plan quirúrgico programado, y
13.2.4.6 Las demás que señala este proyecto de Norma.
13.2.5 Para la selección de pacientes candidatos a disposición de sangre y productos sanguíneos para uso autólogo mediante procedimientos de depósito previo, se deberá hacer una valoración que permita excluir a las personas que presenten cualquiera de las siguientes condiciones:
13.2.5.1 Niños con peso inferior a 10 kg;
13.2.5.2 Padecimientos crónicos con respuesta medular hematopoyética insuficiente;
13.2.5.3 Enfermedad de células falciformes;
13.2.5.4 Enfermedad cardiaca en situación de inestabilidad, tales como:
13.2.5.4.1 Angina inestable;
13.2.5.4.2 Infarto al miocardio ocurrido en los últimos 6 meses;
13.2.5.4.3 Estenosis de las arterias coronarias;
13.2.5.4.4 Cardiopatías cianógenas;
13.2.5.4.5 Hipertensión arterial descontrolada, y
Cualquier otra condición cardiaca, solo previa valoración y autorización escrita por un cardiólogo;
13.2.5.5 Evento vascular cerebral ocurrido en los últimos seis meses. Si el evento ocurrió en un lapso mayor a seis meses, sólo que se cuente con la valoración y autorización escrita por un neurólogo;
13.2.5.6 Neuropatías, tales como: enfermedad cerebrovascular, antecedente de convulsiones o epilepsia, salvo valoración y autorización escrita por un neurólogo;
13.2.5.7 Toxemia gravídica moderada o grave;
13.2.5.8 Infección bacteriana aguda;
13.2.5.9 Reactividad o positividad en cualquiera de los marcadores de infección que se indican a continuación:
13.2.5.9.1 Antígeno de superficie del virus B de la hepatitis;
13.2.5.9.2 Anticuerpos contra el virus C de la hepatitis;
13.2.5.9.3 Anticuerpos contra el virus de la inmunodeficiencia humana, tipos 1 o 2, y
13.2.5.9.4 Anticuerpos contra Trypanosoma cruzi;
13.2.5.10 Cuando la persona tenga antecedente de la presencia de anticuerpos contra el virus HTLV-I o II;
13.2.5.11 Cuando la persona tenga valores de hemoglobina inferiores a 10 g/dL, y
13.2.5.12 De pretender efectuar la extracción mediante eritroaféresis de doble colecta, se excluirán de este procedimiento los candidatos siguientes:
13.2.5.12.1 Las personas que tengan un peso corporal inferior a 70 kg, en ausencia de obesidad (véase la Guía Nacional de Criterios para la Elección de Donantes de Sangre y sus componentes sanguíneos para el uso terapéutico, en su versión más actual, publicada por el Centro Nacional de Transfusión Sanguínea disponible en https://www.gob.mx/cnts/documentos/recursos-guias-y-documentos-en-medicina-transfusional ).
13.2.5.12.2 Las personas que tengan un volumen sanguíneo calculado inferior a cinco litros, y
13.2.5.12.3 Las personas que tengan una cifra de hemoglobina menor a 140 g/L y de hematocrito menor a 42 %.
13.2.6 En cualquier condición en la que los criterios de selección no sean aplicables, el Banco de Sangre o el Servicio de Transfusión Hospitalario deberá contar con reglas específicas, señaladas en los procedimientos normalizados de operación y aprobadas por el responsable sanitario y por el médico que indique el procedimiento.
13.2.7 Recomendaciones adicionales para efectuar un programa de disposición de sangre para uso autólogo mediante depósito previo:
13.2.7.1 En niños con peso corporal entre 10 y 20 kg, deberá hacerse remplazo volumétrico con soluciones;
13.2.7.2 Deberán extremarse los cuidados en personas mayores de 70 años;
13.2.7.3 Durante la extracción sanguínea en embarazadas se deberá vigilar estrechamente la aparición de contracciones uterinas y la frecuencia cardiaca fetal, y
13.2.7.4 En pacientes con valores de hemoglobina entre 10-11 g/dL (100 y 110 g/L), deberá valorarse la realización del procedimiento de autodonación, de acuerdo con el número de unidades programadas y a la etiología de la anemia.
13.2.8 Los Bancos de Sangre, Centros de Colecta y Servicios de Transfusión Hospitalaria, deberán contar con procedimientos normalizados de operación que describan cómo prevenir, tratar y registrar las reacciones adversas que pueden tener los donantes sometidos a procedimientos transfusión autóloga mediante procedimientos de depósito previo.
13.2.9 Al igual que en la donación alogénica, los efectos adversos relacionados con las extracciones para uso autólogo, deberán notificarse al Centro Nacional de la Transfusión Sanguínea de conformidad con los formatos que para ello establezca o a través de medios electrónicos y al comité de medicina transfusional que el establecimiento tuviese.
13.2.10 La extracción de sangre para uso autólogo mediante el procedimiento de depósito previo se deberá realizar bajo las mismas condiciones y procedimientos que para la extracción de sangre para uso alogénico (véanse los subincisos que conforman el inciso 8.1 de este proyecto de Norma).
13.2.10.1 En cada extracción de sangre total o de eritrocitos mediante aféresis, el volumen obtenido en cada flebotomía no deberá exceder de:
13.2.10.1.1 El 10 % del volumen sanguíneo total, en pacientes de ocho años o menores, y
13.2.10.1.2 El 13 % del volumen sanguíneo total, en pacientes mayores de ocho años.
13.2.10.2 El médico tratante juntamente con el médico responsable del Banco de Sangre, Centro de Colecta o del Servicio de Transfusión Hospitalario establecerán la frecuencia y el número de las extracciones de forma individualizada para cada paciente. No es aconsejable que el intervalo entre extracciones de sangre total o de eritroaféresis sea menor a 72 horas.
13.2.10.3 El intervalo mínimo entre la última extracción de sangre o de una eritroaféresis y la fecha programada para la cirugía o la transfusión deberá ser de 72 horas.
13.2.11 A todo donante autólogo por procedimientos de depósito previo, se le deberán practicar las determinaciones analíticas que se indican a continuación, observando las metodologías correspondientes señaladas en el capítulo 10 denominado "Determinaciones analíticas" de este proyecto de Norma.
13.2.11.1 Pruebas para la detección de:
13.2.11.1.2 Treponema pallidum;
13.2.11.1.3 Virus B de la hepatitis;
13.2.11.1.4 Virus C de la hepatitis;
13.2.11.1.5 Virus de la inmunodeficiencia humana tipos 1 y 2, y
13.2.11.1.5 Trypanosoma cruzi;
13.2.11.2 Hemoclasificación ABO y Rh (D);
13.2.11.3 En aquellos que tengan antecedentes propiciadores de aloinmunización se hará investigación de anticuerpos irregulares de importancia clínica, y
13.2.11.4 Las demás que determine el responsable sanitario del Banco de Sangre o del Servicio de Transfusión Hospitalario por razón de la situación epidemiológica de la región geográfica donde se encuentra el establecimiento o de la de procedencia del donante, de sus antecedentes personales o de sus factores de riesgo para adquirir enfermedades infecciosas.
13.2.12 Las pruebas referidas en el inciso anterior deberán realizarse en las muestras sanguíneas obtenidas durante la primera extracción de sangre.
13.2.13 A cualquier unidad con resultado reactivo o positivo en las pruebas de detección de los agentes transmisibles por transfusión deberá dársele destino final y suspender el programa de extracciones para autodonación.
13.2.14 En lo que se refiere al procesamiento, conservación y vigencia de las unidades de sangre y productos sanguíneos obtenidas en un procedimiento de depósito previo, se observarán las disposiciones que señala el capítulo 9 denominado "Procesamiento, conservación, vigencia y control de calidad de las unidades de sangre y productos sanguíneos" de este proyecto de Norma.
13.2.15 Los Servicios de Sangre, deberán contar con mecanismos que garanticen el uso autólogo exclusivo de las unidades; para ello, se deberán mantener bajo estricta custodia, preferentemente separadas del resto de las unidades.
13.2.16 La etiqueta de las unidades de sangre o productos sanguíneos para uso en transfusión autóloga, deberá tener la información siguiente:
13.2.16.1 Nombre del Servicio de Sangre procesador;
13.2.16.2 Las frases siguientes:
13.2.16.2.1 "DONACIÓN AUTÓLOGA"
13.2.16.2.2 "ESTRICTAMENTE RESERVADA PARA": seguida del nombre del paciente, la fecha de su nacimiento y su número de expediente o registro;
13.2.16.3 Nombre del componente sanguíneo de que se trate;
13.2.16.4 La identificación numérica o alfanumérica exclusiva de la unidad;
13.2.16.5 Nombre de la solución anticoagulante o, en su caso, el nombre y volumen de la solución aditiva;
13.2.16.6 Resultado de las pruebas de detección de los agentes infecciosos;
13.2.16.7 Grupo sanguíneo ABO y Rh (D). La presencia o ausencia del antígeno Rh (D) se señalará con letras mayúsculas: "POSITIVO" o "NEGATIVO", o con los símbolos o siglas "+" o "NEG", respectivamente;
13.2.16.8 Volumen aproximado de la unidad;
13.2.16.9 El rango de temperatura en que debe conservarse;
13.2.16.10 Fechas de extracción y de caducidad, y
13.2.16.11 Los señalamientos siguientes:
13.12.16.11.1 No mezclarse con medicamentos, y
13.12.16.11.2 Transfundirse a través de un filtro de 170-200 micras (µm).
13.2.17 Los Servicios de Sangre y los establecimientos para la atención médica, deberán contar con un sistema que permita el conocimiento de que el paciente tiene sangre autóloga disponible, con el fin de evitar transfusiones alogénicas innecesarias.
13.2.18 A las unidades de sangre o productos sanguíneos que no se hubiesen transfundido en la cirugía programada para la cual fueron recolectadas, se les dará destino final o, en su caso, se conservarán en congelación para cubrir posibles requerimientos transfusionales futuros del propio donante.
14 Solicitudes de transfusión, suministro y recepción, traslado y readmisión de unidades de sangre y productos sanguíneos
14.1 Solicitudes de transfusión:
14.1.1 Los Servicios de Sangre deberán tener los Procedimientos Normalizados de Operación para que las solicitudes de unidades o mezclas de productos sanguíneos para uso transfusional sean llenadas apropiadamente y que se mantengan registros de las solicitudes.
14.1.2 Toda solicitud para el suministro de unidades o mezclas de sus componentes deberá contener información suficiente para la identificación del receptor, de su diagnóstico y, los antecedentes de importancia para efectos transfusionales (véase inciso 20.3.4.6 de este proyecto de Norma).
14.1.3 Las solicitudes con información ilegible o discordante con los datos de identificación de la muestra no deberán ser aceptadas por el Banco de Sangre o el Servicio de Transfusión Hospitalario. En caso de formatos de solicitud con información faltante, deberá corroborarse con el solicitante.
14.1.4 Se aceptarán peticiones por vía telefónica o por medios electrónicos mientras proporcionen la información completa. Las solicitudes por vía telefónica deberán ser avaladas por el responsable sanitario del servicio de sangre, con el envío ulterior del formato de solicitud adecuadamente llenado.
14.1.5 Tratándose de solicitudes telefónicas o por medios electrónicos, el personal de los Servicios de Sangre que las reciba las solicitudes deberá llevar un registro escrito que incluya:
14.1.5.1 Fecha y hora de recepción de la solicitud, y
14.1.5.2 Nombre del médico que indica la transfusión y de la persona que realiza la llamada.
14.1.6 Para que los Servicios de Sangre suministren unidades o mezclas de productos sanguíneos por una petición urgente, el médico solicitante deberá justificar Que la situación es de suficiente apremio como para transfundir sin las pruebas de compatibilidad completas.
14.1.7 Para las urgencias transfusionales los Servicios de Sangre deberán contar con procedimientos escritos para suministrar sin dilación unidades de sangre o productos sanguíneos observando las disposiciones que señala el inciso 12.6 de este proyecto de Norma.
14.2 Suministro y recepción de productos sanguíneos.
14.2.1 Antes de suministrar alguna unidad para uso terapéutico, el personal asignado para el suministro de unidades del Servicio de Sangre deberá contar con procedimientos normalizados de operación y validados que permitan la correcta identificación de la unidad a transfundir, receptor y que la unidad cuenta con todos los estudios necesarios antes de su transfusión.
14.3 Los establecimientos para la atención médica deberán implementar mecanismos que aseguren que toda unidad de sangre o componente sanguíneo procedente de otro establecimiento ingresen a través del Servicio de Sangre del establecimiento receptor.
14.4 El personal de un Servicio de Sangre que reciba unidades de sangre y productos sanguíneos procedentes de otros establecimientos similares, deberá observar lo siguiente:
14.4.1 Confirmará su procedencia y que estén adecuadamente identificadas;
14.4.2 Verificará su aspecto físico, condiciones de envío y embalaje (véase inciso 14 .5.2 de este proyecto de Norma);
14.4.3 Se realizará el registro de la temperatura al momento de su recepción;
14.4.4 En su caso, constatará si se realizaron las pruebas de compatibilidad sanguínea de acuerdo con el componente de que se trate;
14.4.5 Registrará las unidades en el libro de ingresos y egresos del establecimiento o su equivalente y en su caso, en los sistemas de cómputo;
14.4.6 Notificará cualquier anormalidad o desviación observada al responsable sanitario del Banco de Sangre, Centro de Procesamiento o Servicio de Transfusión Hospitalario que suministra las unidades. Haciendo el registro correspondiente a la anormalidad o desviación, y
14.4.7 El responsable sanitario o el personal asignado del Banco de Sangre o del Servicio de Transfusión Hospitalario del establecimiento receptor indicará si son aptas o no para uso transfusional, o bien, para su destino final.
14.5 Traslado de unidades de sangre y productos sanguíneos.
14.5.1 Los Servicios de Sangre que hagan algún envío de unidades de sangre o productos sanguíneos, serán responsables del embalaje, conservación, monitoreo y transporte de las unidades, para que su traslado se realice de manera adecuada, bajo condiciones que preservan la integridad y las propiedades terapéuticas del componente sanguíneo de que se trate.
14.5.2 Para el traslado de unidades de sangre y productos sanguíneos de un establecimiento a otro, será aplicativo lo que se indica a continuación:
14.5.2.1 Los Servicios de Sangre proporcionarán al transportista la capacitación adecuada y las instrucciones necesarias para un traslado adecuado;
14.5.2.2 Se colocarán en contenedores o cajas de transporte verificadas, preferentemente de material plástico, que sean herméticas, termoaislantes y lavables y que aseguren que la temperatura interior se mantenga en los rangos adecuados de acuerdo con el componente que se pretende transportar. El tiempo de traslado de las unidades dependerá de la capacidad de las cajas de mantener el rango de temperatura indicado;
14.5.2.3 Las unidades se colocarán en las cajas o contenedores de traslado de forma que se minimicen daños por movimientos violentos o por el contacto directo con los refrigerantes, especialmente cuando se trate de unidades celulares en estado líquido. De emplearse cajas diseñadas específicamente para el traslado de unidades, éstas deberán contar con certificado de validación del fabricante o el proveedor y deberán cumplir con las especificaciones mínimas que señala la Organización Mundial de la Salud (OMS);
14.5.2.4 Debe de contar con un registro de embalaje, con el que quede asentado quién realizó y supervisó el mismo. El registro debe de contener como mínimo la siguiente información:
14.5.2.5 Fecha y hora de inicio de embalaje.
14.5.2.6 Fecha y hora de termino de embalaje.
14.5.2.7 Temperatura en la que se encuentra la unidad una vez colocada en el contenedor, termo o hielera. (Realizar la toma de temperatura con un instrumento calibrado).
14.5.2.8 Número de serie y tipo de termómetro o dispositivo calibrado que se colocó para la medición y trazabilidad de la temperatura.
14.5.2.9 Si cumple o no con el armado o embalaje con base a la calificación o validación de proveedor o fabricante.
14.5.2.10 Firma o iniciales de quién realizó y supervisó.
14.5.2.11 Se tomarán las medidas necesarias a fin de que las unidades se mantengan durante el traslado dentro de los rangos de temperatura que se indican a continuación:
14.5.2.12 Entre +2°C y +10° C, para sangre total o reconstituida, concentrado de eritrocitos y concentrados de eritrocitos descongelados y resuspendidos y plasmas o crioprecipitados en estado líquido;
14.5.2.13 Entre +20° C y +24° C o a temperaturas lo más cercanas a este rango, para preparados con plaquetas;
14.5.2.14 A temperaturas que garanticen el estado de congelación (-20º C a -25ºC) para los plasmas, crioprecipitados, mezclas de crioprecipitados, plaquetas o concentrados de eritrocitos congelados;
14.5.2.15 Entre +20º C y +24º C o a temperaturas lo más cercanas a este rango para unidades de granulocitos;
14.5.2.16 Al arribo de los productos sanguíneos se colocarán en el interior de los equipos adecuados de conservación, a menos que fuesen a transfundirse inmediatamente;
14.5.2.17 Se emplearán dispositivos calibrados que permitan verificar la trazabilidad de la temperatura de conservación durante el traslado para asegurar la conservación óptima;
14.5.2.18 Los contenedores o cajas de transporte con unidades no se colocarán en lugares donde puedan presentarse temperaturas extremas, tales como, los compartimentos de carga de automóviles, autobuses o aviones, a menos que se empleen cajas de transporte específicas para unidades, previendo que la temperatura exterior no fuese a sobrepasar los límites de capacidad de estas cajas para mantener la temperatura en su interior.
14.5.2.19 En transporte aéreo, las unidades de sangre y productos sanguíneos en estado líquido se trasladarán en cabinas presurizadas;
14.5.2.20 El tiempo máximo de traslado de unidades celulares o mezclas de éstas en estado líquido, no deberá exceder de 24 horas, y
14.5.2.21 El tiempo máximo de traslado de plasmas en estado líquido será aquel que permita que las unidades sean transfundidas en un intervalo que no exceda de 24 horas a partir de su descongelamiento y en caso de crioprecipitados, que no exceda de 4 horas.
14.5.3 Para el traslado y recepción de unidades de sangre y productos sanguíneos de algún Servicio de Sangre del cual depende, se observará lo siguiente:
14.5.3.1 El Centro de Colecta dará instrucciones precisas al personal que vaya a efectuar el traslado para que los componentes sean entregados a la brevedad al Banco de Sangre;
14.5.3.2 Las unidades se colocarán en contenedores herméticos y termoaislantes o en cajas de transporte específicamente diseñadas para este fin, en condiciones que permitan el mantenimiento de los rangos de temperatura que se describe a continuación:
14.5.3.2.1 Entre +2° y +10° C, para sangre total o reconstituida, concentrado de eritrocitos y concentrados de eritrocitos descongelados y resuspendidos y plasmas o crioprecipitados en estado líquido;
14.5.3.2.2 Entre +20° y +24° C o a temperaturas lo más cercanas a este rango, para preparados con plaquetas;
14.5.3.2.3 A temperaturas de al menos -20º C que garanticen el estado de congelación para los plasmas, crioprecipitados, mezclas de crioprecipitados, plaquetas o concentrados de eritrocitos congelados;
14.5.3.2.4 Entre +20º C y +24º C o a temperaturas lo más cercanas a este rango para unidades de granulocitos;
14.5.3.3 La remesa se acompañará del impreso referido en el inciso 20.3.4.5 de este proyecto de Norma, y
14.5.3.4 Al momento de la recepción, el personal que reciba las unidades verificará que las unidades vengan adecuadamente identificadas, su estado físico, y se realizará el registro de la temperatura; la medición de la misma, deberá realizarse mediante un dispositivo que permita el seguimiento de los cambios de temperatura al menos cada 10 minutos, durante todo el traslado de los productos sanguíneos, el cual deberá contar con calibración vigente.
14.5.4 Para el traslado y recepción de unidades de sangre y productos sanguíneos de algún Servicio de Sangre a establecimientos similares o un servicio clínico ubicado en el mismo establecimiento para la atención médica, se observará lo siguiente:
14.5.4.1 El Banco de Sangre, Centro de Colecta, Centro de Distribución de Sangre y Productos Sanguíneos, el Servicio de Transfusión Hospitalario o el Centro de Procesamiento, dará capacitación e instrucciones precisas al personal que vaya a efectuar el traslado para que los componentes sean entregados a la brevedad a los servicios clínicos solicitantes;
14.5.4.2 Las unidades se colocarán en contenedores o cajas de transporte herméticos y termoaislantes, en condiciones que permitan el mantenimiento de los rangos de temperatura referidos en el inciso 14.5.2.11;
14.5.4.3 Al momento de la recepción, el personal que reciba las unidades o mezclas registrará en una libreta específica la información que señala el inciso 20.3.48 de este proyecto de Norma;
14.5.4.4 El personal asignado de los servicios clínicos transfundirá a la brevedad posible las unidades asignadas a cada paciente, previa verificación de:
14.5.4.4.1 La compatibilidad ABO y Rh (D), cuando el componente lo requiera, y
14.5.4.4.2 Que se hubieran efectuado las determinaciones analíticas obligatorias, incluyendo las pruebas cruzadas, salvo en los casos urgentes, de conformidad a lo que establece este proyecto de Norma, y
14.5.4.5 Ante cualquier duda, antes del inicio de la transfusión, el personal de los servicios clínicos la resolverá con el Servicio de Sangre correspondiente.
14.6 Retorno al Servicio de Sangre correspondientes de productos sanguíneos no transfundidos.
14.6.1 Los Servicios de Sangre establecerán un procedimiento para la readmisión de unidades o mezclas previamente suministradas. Las unidades de sangre o productos sanguíneos o mezclas de productos sanguíneos podrán admitirse de nuevo a las reservas del Banco de Sangre o Servicio de Transfusión Hospitalario, siempre y cuando se cumplan los requisitos siguientes:
14.6.1.1 Que las unidades o mezclas conserven el sistema cerrado (bolsas no abiertas o "picadas");
14.6.1.2 Que el componente se haya mantenido continuamente a la temperatura adecuada para su conservación, y
14.6.1.3 Que no se observen cambios de coloración, presencia de hemólisis o cualquier otro cambio físico.
14.6.1.4 Sean enviadas con las condiciones de embalaje que permitan la conservación óptima
14.6.2 Los Servicios de Sangre podrán recibir las unidades que no vengan en las condiciones que establece el inciso anterior únicamente para darles destino final.
14.6.3 Los Servicios de Sangre deberán documentar la identificación correcta de la unidad o mezcla, fecha y hora del suministro y retorno de cada componente sanguíneo, así como los resultados de su inspección y, en su caso, la aceptación o destino final del mismo.
15 Transfusión de unidades y reacciones adversas a la transfusión
15.1 La indicación de una transfusión será responsabilidad del médico tratante o del médico que la prescriba.
15.2 El médico tratante deberá limitar el uso terapéutico de la sangre y productos sanguíneos a los casos en que se reúnan las condiciones siguientes:
15.2.1 Cuando el receptor tenga un padecimiento que no sea susceptible de corregirse por otros métodos terapéuticos, y
15.2.2 Cuando el beneficio terapéutico predecible supere los riesgos inherentes.
15.3 Para mejor indicación y uso terapéutico de la sangre y productos sanguíneos, deberá apegarse a las recomendaciones del documento referenciado en el inciso 22.10 de este proyecto de Norma).
15.4 El médico tratante será el responsable de la indicación de las transfusiones, mismas que podrán aplicarse y supervisarse por otros trabajadores de la salud, tales como médicos o personal de enfermería, capacitados en la aplicación y vigilancia de las transfusiones.
15.5 La identificación del paciente que va a recibir una transfusión deberá hacerse por dos profesionales de la salud, quienes verificarán con especial atención lo siguiente:
15.5.1 La identidad correcta del receptor, mediante las acciones siguientes:
15.5.1.1 Corroboración verbal cuando esto sea posible, así como revisión del nombre anotado en la pulsera de identificación del paciente, y
15.5.1.2 A través de los registros del expediente clínico;
15.5.2 La concordancia de los datos contenidos en la solicitud con los de la etiqueta de la unidad que se va a transfundir y el marbete que la acompaña, en lo relativo al número exclusivo de la unidad, el grupo AB0 y Rh y, cuando el componente lo requiera, las pruebas cruzadas de compatibilidad efectuadas, y
15.5.3 Que la etiqueta de la unidad consigne los demás resultados de las determinaciones analíticas obligatorias que establece este proyecto de Norma.
En caso de haber discrepancia o duda en materia de lo referido en este apartado, el personal de salud deberá diferir la transfusión hasta su esclarecimiento.
15.6 Los Servicios de Sangre y los servicios clínicos de los establecimientos para la atención médica que en sus instalaciones apliquen transfusiones, deberán contar con registros de las transfusiones aplicadas que contendrán como mínimo la información que señala el inciso 20.3.4.9 de este proyecto de Norma.
15.7 El médico que indique una transfusión deberá registrar o supervisar que el personal que la aplique registre en el expediente clínico del receptor las transfusiones que se hayan aplicado, anotando como mínimo, la información que señala el inciso 20.3.4.10 de este proyecto de Norma.
15.8 El responsable sanitario de un Banco de Sangre o de un Servicio de Transfusión Hospitalario, deberá procurar que se realicen los registros a que se refiere el apartado que antecede.
15.9 Las transfusiones ambulatorias se podrán aplicar en el Servicio de Sangre o en un servicio de atención médica, que cuente con personal capacitado y los recursos necesarios para atender cualquier evento o reacción adversa a la transfusión.
15.10 El acto transfusional no deberá exceder de tres horas para cada unidad de concentrado de eritrocitos o de sangre. Las unidades de crioprecipitados o de plaquetas deberán transfundirse tan rápido como la vía intravenosa lo permita.
15.11 Antes o durante una transfusión no deberán agregarse medicamentos o fármacos a las unidades de sangre o de sus componentes, aún aquéllos que sean destinados para uso intravenoso, con excepción de solución de cloruro de sodio al 0.9% estéril.
15.12 Las unidades de sangre o de sus componentes deberán mantenerse en condiciones de conservación apropiadas y óptimas hasta el momento de su aplicación terapéutica. Las unidades de sangre y productos sanguíneos en estado líquido no deberán ser sometidas a ningún tipo de calentamiento previo a la transfusión, salvo en los casos siguientes:
15.12.1 Cuando se requiera administrar 15 mL o más por minuto;
15.12.2 En exsanguineotransfusión, y
15.12.3 Cuando el receptor sea portador de crioaglutininas.
En cualquiera de estos casos, en el momento previo inmediato a la transfusión, las unidades podrán someterse a calentamiento a una temperatura que no exceda de +38° C o bien, durante el acto transfusional mediante el pasaje de la sangre por un equipo específico con control de temperatura, termómetro visible y sistema de alarma.
15.13 Para la transfusión de unidades de sangre y productos sanguíneos se deberán utilizar equipos con filtro de 170 a 200 micras (µm) estériles y libres de pirógenos, capaces de retener microagregados, los que se emplearán individualmente y se desecharán en el momento que ocurra cualquiera de lo siguiente:
15.13.1 Cuando tengan cuatro horas de uso, o
15.13.2 Al haber transfundido cuatro unidades.
15.14 Ante síntomas o signos de una reacción transfusional, el médico tratante o el personal de salud deberá interrumpir inmediatamente la transfusión e iniciar el protocolo, en lo que se esclarece su causa y se investiga un posible error en la identificación del receptor y de la unidad. Deberá seguir los siguientes diagramas para el abordaje clínico y de laboratorio de Reacciones Adversas a la Transfusión.
Figura 4
Diagrama de flujo para el abordaje clínico de Reacciones Adversas a la Transfusión.
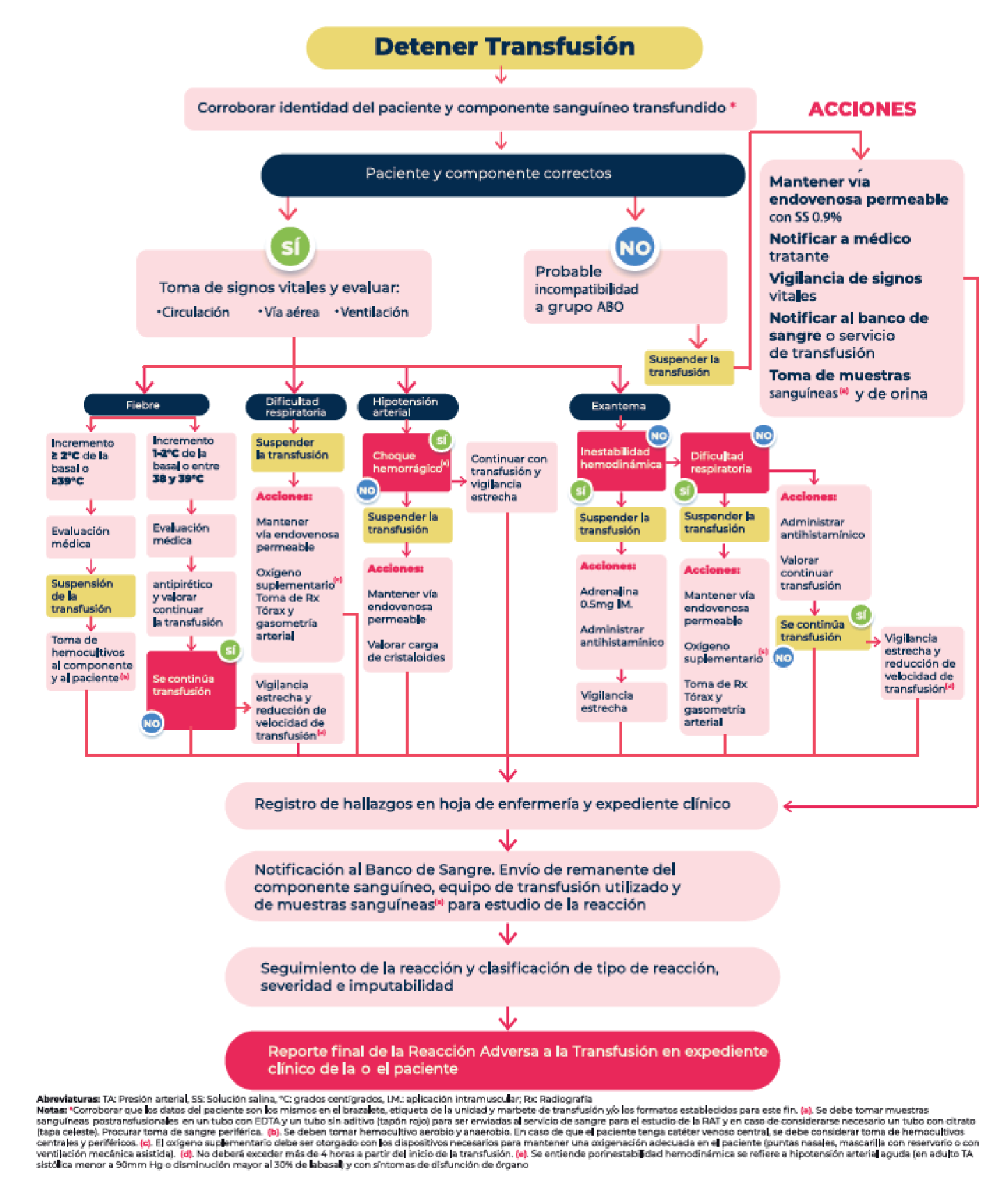
Figura 5
Diagrama de flujo para el abordaje de laboratorio de Reacciones Adversas a la Transfusión.

15.15 El médico que atienda a un paciente que ha recibido una transfusión, deberá evaluar de inmediato cualquier aparente reacción transfusional y adoptará las medidas que estime necesarias, conforme a los procedimientos establecidos. (consúltese el documento referenciado en el inciso 22.10 de este proyecto de Norma).
15.16 El médico o el personal de salud que atienda al paciente deberá notificar al Banco de Sangre o al Servicio de Transfusión Hospitalario y, en su caso, al comité de medicina transfusional que el establecimiento de atención médica tuviese, todas las aparentes reacciones adversas que se hubiesen presentado. Deberá quedar asentado en expediente clínico del paciente, el tipo de reacción, así como la nota médica donde se tenga el resultado del análisis del tipo de reacción, y el seguimiento por el médico tratante.
15.17 Ante la sospecha de una reacción o efecto adverso imputable a una transfusión, el servicio clínico del establecimiento de atención médica deberá enviar en un tiempo máximo de 30 minutos al Servicio de Sangre correspondiente, el marbete a que se refiere el inciso 20.3.4.7 de este proyecto de Norma, a fin de se hagan las determinaciones analíticas necesarias para esclarecer las causas, acompañado de lo que se indica a continuación:
15.17.1 Muestras postransfusionales del receptor obtenidas con y sin anticoagulante, adecuadamente recolectadas para evitar hemólisis y apropiadamente identificadas, y
15.17.2 La unidad que se estaba transfundiendo, aunque no contuviese residuo, así como el equipo de transfusión y las soluciones intravenosas que se estuvieran administrando.
15.18 La sobrecarga circulatoria también requiere ser informada al Banco de Sangre o Servicio de Transfusión Hospitalario, más no será necesaria su intervención para la evaluación del evento.
15.19 En caso de una reacción transfusional inmediata, el Banco de Sangre o, en su caso, el Servicio de Transfusión Hospitalario deberá llevar a cabo simultánea y comparativamente los procedimientos y pruebas de laboratorio que permitan resolver la causa de la reacción adversa, consultando la técnica que especifique su manual de procedimientos de resolución de reacciones adversas, de conformidad con el inciso 15.14 de este proyecto de Norma.
15.20 Ante la sospecha de una reacción o efecto adverso imputable a una transfusión grave, el Comité de Medicina Transfusional deberá sesionar en un periodo máximo de 24 horas, para realizar el reporte de Hemovigilancia y notificar al CNTS, así como enviar las acciones preventivas, correctivas y de mejora, generadas para dar atención al evento.
16 Evaluación de la conformidad y control de calidad
16.1 Disposiciones comunes:
16.1.1 Los Bancos de Sangre, Centros de Calificación Biológica, Centros de Procesamiento y los Servicios de Transfusión Hospitalario, deberán participar en programas calendarizados de evaluación de la conformidad que realizan las dependencias competentes y por los que determine la Secretaría de Salud o por los organismos de certificación y acreditación conforme a lo señalado en la Ley de Infraestructura de la Calidad.
Se exceptúan de lo anterior los centros de colecta por tratarse dependencias de un banco de sangre y funcionar bajo su responsabilidad y aquellos servicios de transfusión hospitalario que únicamente reciban unidades de sangre y productos sanguíneos previamente evaluadas, analizadas y listas para su aplicación terapéutica en un determinado paciente, pero deben contar con procedimientos para conservación de unidades de productos sanguíneos y de establecimiento y seguimiento de un programa de hemovigilancia, así como aquellos destinados a indicar como realizar la transfusión y el destino final de las unidades.
16.1.2 Los Bancos de Sangre, Centros de Colecta y Centros de Procesamiento de Sangre deben establecer a través del uso de herramientas estadísticas el control estadístico de proceso, para monitorear y darle seguimiento a los resultados de control de calidad en las áreas descritas a continuación:
16.1.2.1 Procesamiento de sangre y productos sanguíneos.
16.1.2.2 Determinación de agentes transmisibles por transfusión.
16.1.2.3 Inmunohematología.
16.1.2.4 Control de calidad de productos sanguíneos;
16.1.2.4.1 Establecimiento de frecuencia de realización de control de calidad.
16.1.2.4.2 Establecimiento de parámetros de calidad (volumen).
16.1.3 Los laboratorios de los Bancos de Sangre, los Centros de Calificación Biológica, los Centros de Procesamiento y en su caso, de los Servicios de Transfusión Hospitalario que realicen determinaciones analíticas, deberán participar en los programas de control de calidad externo que aplica el Centro Nacional de la Transfusión Sanguínea de conformidad con lo que se indica a continuación:
16.1.3.1 Participar en los programas de control de calidad externo en las modalidades siguientes:
16.1.3.1.1 Pruebas para la detección de agentes infecciosos transmisibles por transfusión (tamizaje serológico y molecular), y
16.1.3.1.2 Pruebas de inmunohematología;
16.1.3.2 Los establecimientos participantes deben enviar los resultados obtenidos por medio de correo electrónico u otros medios que determine el Centro Nacional de la Transfusión Sanguínea;
16.1.3.3 El pago de cuota de recuperación que establezca el Centro Nacional de la Transfusión Sanguínea para cada control de calidad y cada lote de evaluación, deberá realizarse a nombre del establecimiento declarado en la licencia sanitaria vigente y con el R.F.C. correspondiente a la persona física o moral propietaria del establecimiento, declarada en la licencia sanitaria.
16.1.3.4 Los establecimientos tendrán un plazo de respuesta que no excederá de veinte días naturales contados a partir de que reciban las muestras por parte del Centro Nacional de la Transfusión Sanguínea;
16.1.3.5 El responsable sanitario de un Banco de Sangre, Servicio de Transfusión Hospitalario o Centro de Calificación Biológica , revisará, acatará y supervisará que se lleven a cabo las indicaciones que hiciere el Centro Nacional de la Transfusión Sanguínea como consecuencia de la aplicación de estos programas, siendo corresponsable la persona física o moral propietaria del establecimiento, declarada en la licencia sanitaria, y
16.1.3.6 Las acciones preventivas y correctivas efectuadas por los establecimientos y sus resultados deberán registrarse y se llevará a cabo un análisis de riesgo enfocado en el impacto de lo detectado.
Para garantizar el buen funcionamiento de los programas de control de calidad externo, en los casos que se considere necesario, los Centros Nacional y Estatales de la Transfusión Sanguínea podrán otorgar asesoría presencial o a distancia a los establecimientos con el fin de aplicar las medidas preventivas o correctivas que resulten necesarias, mismas que también deberán documentarse.
16.1.4 Los Bancos de Sangre, Centros de Procesamiento Servicios de Transfusión Hospitalario o Centros de Calificación Biológica que realicen determinaciones analíticas deberán participar en un control de calidad externo adicional al referido en el inciso anterior.
16.1.5 Los proveedores de las muestras para el control externo de la calidad deberán contar con el reconocimiento de su capacidad técnica y confiabilidad por una entidad de acreditación como proveedor de ensayos de aptitud.
16.1.6 Los servicios de sangre, deberán contar con sistemas de control de calidad interno que incluya todos los procedimientos, desde la selección del donante hasta la transfusión o destino final de las unidades de sangre y productos sanguíneos.
16.1.7 Para asegurar que todos los productos sanguíneos reúnan los requisitos de calidad y seguridad y que los métodos de procesamiento y estudio funcionen como es esperado, todos los Servicios de Sangre, de acuerdo las funciones y actividades que desempeñan, deberán contar con el sistema de gestión de calidad referido en capítulo 5 denominado "Disposiciones generales " de este proyecto de Norma el que deberá incluir, entre otros:
16.1.7.1 Programas de evaluación interna de equipos, instrumentos de medición, pruebas, técnicas y reactivos, que serán efectuados en el propio establecimiento de manera planeada y programada a intervalos previstos y llevados a cabo por personal calificado independiente al personal operativo. En los establecimientos con capacidad instalada reducida, estos programas podrán ser aplicados por el propio personal operativo. La evaluación interna, deberá abarcar las etapas preanalítica, analítica y postanalítica, y
16.1.7.2 Registros de todas las acciones realizadas a efecto del cumplimiento de las disposiciones de este capítulo, así como los incidentes de trascendencia relacionados con los equipos, instrumentos de medición, pruebas, reactivos y técnicas, asimismo, contarán con instrucciones precisas para el tipo de incidente y de cómo actuar en cada caso.
16.1.8 Todos los Servicios de Sangre, en el ámbito de las funciones que desempeñan, deberán disponer de procedimientos escritos para controlar y realizar el seguimiento del equipamiento:
16.1.9 Incluir el equipo de reciente instalación, equipo descompuesto, equipo propio y en comodato.
16.1.10 Mantener registro del estado que guarda todo el equipamiento.
16.1.11 Señalar si ha recibido mantenimiento correctivo o preventivo.
16.1.12 Los equipos defectuosos o pendientes de reparación deberán retirarse e identificarse de forma clara.
16.2 Los controles, las responsabilidades y el responsable del manejo de una desviación deben estar definidos en el procedimiento de que se trate.
16.3 Los establecimientos deben mantener registros de las desviaciones, de su naturaleza, de las acciones correctivas tomadas y de los resultados de las acciones realizadas.
16.4 Al corregir una desviación el establecimiento hará una nueva verificación para demostrar que las acciones correctivas dan cumplimiento a los requisitos establecidos en el Sistema de Gestión de Calidad.
16.5 El responsable sanitario y el personal implicado deberán revisar las desviaciones, las acciones correctivas tomadas y los resultados, a fin de establecer acciones que prevengan su aparición. De igual manera deberán determinar las acciones para prevenir las desviaciones potenciales.
16.6 Control de reactivos.
16.6.1 Cada lote nuevo de reactivos deberá someterse a un proceso interno de inspección, a fin de verificar si cumplen o no con las características requeridas por el laboratorio del establecimiento y las indicadas por el fabricante.
16.6.2 El proceso interno de inspección a que se refiere el inciso anterior deberá incluir como mínimo lo siguiente:
16.6.2.1 Comprobar que el estado del embalaje sea adecuado;
16.6.2.2 Las condiciones de conservación al momento de la recepción;
16.6.2.3 Verificar aspecto físico;
16.6.2.4 Lote y caducidad, y
16.6.2.5 Concordancia de lo descrito en el inserto y el contenido del juego de reactivos.
16.6.3 Debe contar con un procedimiento por escrito que describa el proceso interno de inspección y que incluya las acciones a realizar cuando no cumple con los requisitos mínimos
16.6.4 Para verificar el funcionamiento adecuado de los reactivos, éstos deberán ser probados en forma regular, empleando muestras representativas de cada lote y con la periodicidad que indica la Tabla 34 de este proyecto de Norma.
Tabla 34
Verificación del funcionamiento de los reactivos
Reactivos |
Criterios para su valoración y aceptación |
Periodicidad de comprobación |
Antisueros hemoclasificadores para determinar grupos sanguíneos AB0 y Rh (D). |
1. Aspecto físico: Ausencia de turbidez, precipitados, partículas, formación de geles en el sobrenadante o cualquier otra anomalía. |
Cada día de uso. |
2. Titulación: se llevará a cabo de conformidad con la Farmacopea de los Estados Unidos Mexicanos (véase el inciso 22.1 de este proyecto de Norma). |
Al estreno del lote con una muestra aleatoria de éste. |
|
4. Especificidad: Se realizará conforme a lo indicado en la Farmacopea de los Estados Unidos Mexicanos (véase inciso 22.1 de este proyecto de Norma). Para los antisueros hemoclasificadores AB0 se usarán células A1, A2, B y 0; para los del Rh se emplearán células R1r y rr. |
||
Eritrocitos grupo: A, B, para hemoclasificación AB0 mediante la prueba inversa (grupo sérico). |
1. Aspecto físico: Ausencia de hemólisis, turbidez, precipitados, partículas, formación de geles en el sobrenadante o cualquier otra anomalía. |
Cada día de uso. |
2. Reactividad/especificidad: Reacciones bien definidas con anti-A, anti-B y, de utilizarse, con anti- A,B. |
||
Antiglobulina humana (prueba de Coombs). |
Especificidad con eritrocitos sensibilizados con lo que se indica a continuación a) Con inmunoglobulina G; b) Con eritrocitos con C3b, C3d o ambos, y c) Con eritrocitos no sensibilizados. |
Cada día de uso. |
Eritrocitos sensibilizados con IgG para el control de la antiglobulina. |
Reacción clara de los eritrocitos a los que se le añade antiglobulina |
Cada día uso. |
Eritrocitos reactivos para el rastreo de anticuerpos irregulares de importancia clínica. |
1. Aspecto físico: Ausencia de hemólisis, turbidez, precipitados, partículas, formación de geles en el sobrenadante o cualquier otra anomalía. |
Con cada lote. |
2. Demostrar la presencia de anticuerpos irregulares en un suero que contenga un anticuerpo con especificidad conocida y deberá probarse. |
||
3. Con control externo |
Semestral. |
|
Antisueros para fenotipo de eritrocitos. |
Reacción positiva con eritrocitos heterocigóticos para el antígeno y reacción negativa con eritrocitos carentes del antígeno. |
Cada día de uso. |
Solución salina isotónica al 0.9%. |
Aspecto físico: Ausencia de partículas, formación de geles, alteraciones de color u otras. |
Cada vez que se utilice. |
Reactivos para las pruebas de detección de agentes transmisibles por transfusión. |
Sensibilidad y especificidad empleando controles conocidos, negativos y débilmente positivos. El control débil positivo no deberá ser mayor de tres veces el valor del punto de corte. |
Establecer la frecuencia de uso con base en la planeación del control de calidad |
16.6.5 Ante cualquier anomalía o desviación de los requisitos señalados en la tabla anterior, los reactivos no deberán utilizarse y, de considerarlo necesario se investigarán las causas y se notificará al responsable sanitario del servicio de sangre, al proveedor, al fabricante.
16.7 Control de las pruebas
16.7.1 En caso de obtener resultados falsos o desviaciones en el control de calidad de las pruebas para la detección de agentes infecciosos y de las pruebas de inmunohematología, el personal del laboratorio deberá identificar si los orígenes de los errores están relacionados con cualquiera de lo siguiente:
16.7.1.1 Incumplimiento del seguimiento de las instrucciones proporcionadas por el fabricante;
16.7.1.2 Desempeño inadecuado de los reactivos;
16.7.1.3 Defectos en la operación de equipos e instrumentos;
16.7.1.4 Cálculos e interpretaciones erróneas de la prueba;
16.7.1.5 Fallas en equipos e instrumentos, y
16.7.1.6 Errores en transcripciones y registros, así como, otros errores humanos.
16.7.2 En las pruebas de tamizaje para la detección de agentes transmisibles por transfusión no moleculares, se deberán introducir los controles de calidad internos que se indican a continuación que permitan verificar su desarrollo correcto:
16.7.2.1 Se sugiere establecer numerales y no letras, se deberá incluir un control positivo débil, adicional al que incluye el fabricante, cuyo valor no excederá de tres veces el punto de corte. Este control podrá ser elaborado por el propio establecimiento utilizando una técnica validada, o bien, adquirido de una fuente confiable, y
16.7.2.2 Deberán aplicar el uso de gráficas de control observando las "reglas de zona".
17 Destino final de las unidades de sangre, productos sanguíneos y de las muestras
17.1 El destino final de las unidades de sangre, productos sanguíneos y muestras de éstos, podrá ser la conservación permanente en serotecas o similares, o bien su desecho en las condiciones sanitarias de conformidad con la norma citada en el inciso 2.20 de este proyecto de Norma.
17.2 El personal de los Bancos de Sangre, Servicios de Transfusión Hospitalarios, servicios clínicos y quirófanos, deberá manejar y dar destino final a las unidades de sangre y productos sanguíneos o a las muestras sanguíneas de manera que minimice la exposición potencial a agentes infecciosos.
17.3 El plasma y otros productos sanguíneos que no fueran a utilizarse con fines transfusionales, podrán utilizarse para fines diagnósticos o de investigación, o bien, destinarse para la fabricación de hemoderivados y otros productos biotecnológicos de aplicación terapéutica, diagnóstica, preventiva o en investigación, de conformidad a lo que establezca la Farmacopea de los Estados Unidos Mexicanos, (FEUM, y ediciones actualizadas posteriores), (véase inciso 22.1de este proyecto de Norma).
17.4 El plasma que vaya a destinarse para la fabricación de hemoderivados deberá reunir los requisitos de calidad necesarios a fin de que resulten inocuos, no patogénicos y las fracciones que se pretendan separar deberán ser funcionales, de conformidad con las disposiciones que establezca la Farmacopea de los Estados Unidos Mexicanos, (FEUM, y ediciones actualizadas posteriores), (véase inciso 22.1 de este proyecto de Norma).
17.5 Las serotecas donde se conservan plasmas o sueros que tengan los Bancos de Sangre, Servicios de Transfusión Hospitalario, Centro de Procesamiento y Centro de Distribución deberán reunir los requisitos siguientes:
17.5.1 Los plasmas o sueros en conservación estarán bajo estricta custodia;
17.5.2 Estarán almacenados por fechas, de manera ordenada y limpia y separados de acuerdo con el uso que se les pretenda dar, y
17.5.3 La temperatura para su conservación será de -18ºC o inferiores. Mientras más bajas sean las temperaturas, mayor será la longevidad y la conservación de las propiedades de lo almacenado.
17.5.4 Una muestra no podrá ser congelada y descongelada por más de dos ocasiones para poder realizar determinaciones analíticas.
17.6 El plasma que sea destinado a fraccionamiento industrial por parte de los bancos de sangre, Servicios de Transfusión Hospitalarios, Centros de Procesamiento o Centros de Distribución, que no cumpla con alguno de los requisitos de la materia prima para elaborar hemoderivados señalados en la FEUM, deberá ser desechado y darse destino final por parte de la planta procesadora de plasma a la que fue entregado, asegurando que se resguarde la trazabilidad, desde el origen y obtención de la sangre total o procedimiento de aféresis, hasta el destino final, de conformidad con la norma citada en el inciso 2.20 de este proyecto de Norma en materia de disposición de residuos peligrosos biológico infecciosos.
18 Comité de medicina transfusional
18.1 Todos los establecimientos que realicen actos de disposición de sangre y productos sanguíneos, deberán contar con un Comité de Medicina Transfusional, el cual se sujetará a las disposiciones que para tal efecto emita la Secretaría de Salud, conforme a lo establecido en el artículo 316 de la LGS.
18.2 El Comité de Medicina Transfusional deberá apegarse a lo establecido en el documento de Recomendaciones para la Conformación, Estructura y Funcionamiento del Comité de Medicina Transfusional en los Servicios de Salud, emitido por el CNTS, en su versión más actual, disponible en: https://www.gob.mx/cnts/documentos/recursos-guias-y-documentos-en-medicina-transfusional :
18.3 El comité deberá elaborar y reportar como mínimo los Indicadores señalados en la tabla 35 de este proyecto de norma, de forma mensual.
Tabla 35
1 |
Tasa de diferimiento de donantes: a) total b) permanente c) temporal |
2 |
Quejas sobre productos sanguíneos |
3 |
Quejas de donantes |
4 |
No conformidades de materiales y equipos |
5 |
Evento adverso grave |
18.4 El comité de medicina transfusional deberá sesionar cuando menos cada tres meses o con mayor frecuencia de considerarse necesario.
18.5 Las actas del Comité tendrán que ser resguardadas en el archivo del servicio de sangre y conservarse por un lapso mínimo de cinco años en archivo activo y cinco años en archivo muerto.
19 Información relativa a la disposición de sangre y productos sanguíneos a la Secretaría de Salud
19.1 Corresponde a los servicios de sangre, en el ámbito de las funciones que se les autorizan, informar a la Secretaría sobre los actos de disposición de sangre o productos sanguíneos que realizan y deberán incluir la notificación de las reacciones o eventos adversos que ocurran asociados a la donación o a la transfusión.
19.2 Los informes y la documentación del comité de medicina transfusional deberán presentarse a través de la plataforma digital: "Sistema Analítico Nacional para Gestionar Reportes de Hemcomponentes" (SANGRHE), o a través del medio oficial que disponga la Secretaría para tal fin.
19.3 Para efectos de lo anterior, los servicios de sangre requerirán de accesos personalizados a la plataforma digital, para lo cual deberán contar con licencia sanitaria y aviso de responsable sanitario vigentes, emitidos por la Comisión Federal contra Riesgos Sanitarios.
19.3.1 También deberán contar con Clave Única de Establecimientos de Salud (CLUES) emitida por la Dirección General de Información en Salud, de conformidad con lo establecido en la norma citada en el inciso 2.10 de este proyecto de norma. El procedimiento específico para presentar la información antes mencionada, lo establecerá la Secretaría a través del medio oficial que disponga para tal fin.
19.4 Los servicios de sangre deberán cumplir con las disposiciones aplicables a la plataforma digital SANGRHE, como instructivos de llenado, manual de políticas, acuerdos y demás documentos emitidos por la Secretaría.
19.5 Es el responsable sanitario de los servicios de sangre, el obligado a presentar dichos informes y documentación del comité de medicina transfusional.
19.6 Los informes y la documentación del comité de medicina transfusional, deberá hacerse llegar a la Secretaría mensualmente dentro de los primeros ocho días naturales del mes siguiente al que se informa, o de la prórroga autorizada, de lo contrario el servicio de sangre estará incumpliendo la normatividad vigente y estará sujeto a sanciones por la autoridad sanitaria competente.
19.7 Los servicios de sangre deberán mantener actualizada la información del establecimiento como correos electrónicos, teléfonos, licencias sanitarias, altas de responsable sanitario, y los que requiera la Secretaría, mediante el procedimiento específico que establezca a través del medio oficial que disponga para tal fin.
19.8 Para fines de información epidemiológica, además del informe relativo a la disposición de sangre y productos sanguíneos, los servicios de sangre deberán notificar a la jurisdicción sanitaria las donaciones en las que se hubiese detectado algún resultado positivo en las pruebas confirmatorias o suplementarias para algún agente infeccioso transmisible por transfusión, de conformidad con lo establecido en la norma citada en el inciso 2.14 de este proyecto de Norma.
20 Procedimientos normalizados de operación, guías, instructivos, documentos y registros
20.1 Disposiciones comunes
20.1.1 Los procedimientos normalizados de operación (PNO), guías o instructivos, así como los demás documentos y registros a que hace referencia este capítulo, que tenga un Banco de Sangre, Centro de Colecta, Centro de Procesamiento de Sangre, Centro de Distribución, Centro de Calificación Biológica y Servicio de Transfusión Hospitalario o, en su caso, un servicio clínico de un establecimiento para la atención médica, estarán adecuadamente identificados, con el nombre del establecimiento y el nombre del documento.
20.1.2 Tratándose de los manuales, PNO, guías o instructivos, deberán contener, además, la versión, número de páginas y el nombre y firmas del o los responsables de quien elaboró, revisó y autorizó, esta última siempre es la del responsable sanitario y redactarlos en español, con lenguaje accesible y de fácil comprensión.
20.1.3 Los documentos referidos en el inciso anterior se podrán mantener en sistemas digitales, impresos o una combinación de éstos; en cualquiera de los casos la información deberá ser equivalente y accesible en todo momento.
20.1.4 El Banco de Sangre, Centro de Colecta, Centro de Procesamiento de Sangre, Centro de Distribución, Centro de Calificación Biológica y Servicio de Transfusión Hospitalario, en el ámbito de las funciones que realizan, deberán disponer de sistemas para la obtención y captura de datos que garanticen la trazabilidad de cada unidad y cualquier componente sanguíneo, desde su extracción hasta su uso terapéutico, su destino final o, en su caso, suministro para la elaboración de hemoderivados, incluyendo todos los pasos intermedios del proceso.
20.1.5 La información relativa a la disposición de sangre y productos sanguíneos contenida en sistemas digitales o documentos impresos se mantendrá adecuadamente resguardada y protegida contra cualquier eventualidad. Los documentos escritos se resguardarán a manera de impedir su deterioro.
20.1.6 El responsable sanitario deberá asegurar que el personal ha firmado la carta de compromiso de confidencialidad de los datos sensibles y confidenciales que se encuentran en los archivos impresos y electrónicos del Banco de Sangre, Centro de Colecta, Centro de Procesamiento de Sangre, Centro de Distribución, Centro de Calificación Biológica y Servicio de Transfusión Hospitalario.
20.1.7 La información relativa a la disposición de sangre y productos sanguíneos estará accesible a las autoridades competentes cuando éstas lo soliciten, incluyendo aquella información considerada confidencial. Sin perjuicio de lo establecido en otras disposiciones jurídicas, se considerará de naturaleza confidencial la historia clínica de los donantes o pacientes, los resultados del proceso de autoexclusión del donante y las determinaciones analíticas para la detección de agentes infecciosos transmisibles por transfusión.
20.1.8 Cuando la información relativa a la disposición de sangre y productos sanguíneos se mantenga en registros digitales, los sistemas y su programación deberán estar validados y tener como mínimo los requisitos siguientes:
20.1.8.1 Los sistemas deberán tener mecanismos de control de acceso que puedan verificarse;
20.1.8.2 Solamente el responsable sanitario del establecimiento podrá asignar al personal que pueda tener acceso a los sistemas;
20.1.8.3 Los mecanismos de control de acceso podrán ser sistemas biométricos, de certificación digital, por medio de claves de usuario y contraseñas o cualquier otro mecanismo;
20.1.8.4 Los mecanismos de control de acceso deberán asegurar accesos personales, únicos e intransferibles, que garanticen el nivel de seguridad de acuerdo con el tipo de aplicación;
20.1.8.5 Capacidad de generar documentos impresos que equivalgan a documentos originales;
20.1.8.6 Capacidad de generar bitácoras detalladas sobre todas las acciones realizadas, que incluyan cuando menos fecha, hora, actividad o proceso, variables afectadas y el nombre del usuario. Asimismo, el sistema permitirá que las bitácoras puedan ser revisadas y acceder a la información previa a las modificaciones o cambios;
20.1.8.7 Capacidad para utilizar la información contenida en el sistema, a través de mecanismos de descarga de la información y procedimientos para su análisis;
20.1.8.8 Deberán estar amparados por documentación explícita que detalle, entre otros:
20.1.8.8.1 Instrucciones de uso y administración;
20.1.8.8.2 Capacidades técnicas del sistema y aspectos sobre la seguridad, integridad y riesgos;
20.1.8.8.3 Definiciones relativas al soporte técnico ante cualquier eventualidad;
20.1.8.8.4 Mecanismos de actualización y desarrollos posteriores, y
20.1.8.8.5 En su caso, requerimientos para su uso de acuerdo con la infraestructura de tecnologías de la información prexistente, y
20.1.8.9 Los demás lineamientos de tecnologías de la información en salud que resulten aplicables.
20.1.9 En relación con los registros que lleve el Banco de Sangre, Centro de Colecta, Centro de Procesamiento de Sangre, Centro de Distribución, Centro de Calificación Biológica y Servicio de Transfusión Hospitalario, se deberá observar lo siguiente:
20.1.9.1 Tendrán procedimientos escritos para llevar los registros que tengan que conservarse y de cómo, dónde y durante cuánto tiempo habrán de mantenerse;
20.1.9.2 Los registros estarán escritos en lenguaje comprensible, uniforme y preciso, evitando, en lo posible, ambigüedades o la inclusión de aspectos opcionales. En sistemas digitales es preferible que el ingreso de la información se haga en campos de selección predeterminados. Tratándose de documentos escritos, las anotaciones deberán ser indelebles, legibles, el registro de actividades debe realizarse a momento de la actividad respetando el orden cronológico y
20.1.9.3 Identificarán a la persona directamente responsable de cada tarea.
20.1.10 El Banco de Sangre, Centro de Colecta, Centro de Procesamiento de Sangre, Centro de Distribución, Centro de Calificación Biológica y Servicio de Transfusión Hospitalario deberán tener registros de las revisiones y, en su caso, aprobaciones de los cambios que en su caso se hagan, en relación con el funcionamiento del establecimiento, a los manuales, procedimientos normalizados de operación, guías, instructivos u otros documentos, así como de los cambios en la metodología para llevar a cabo los registros.
20.1.11 Se deberá contar con la aprobación del responsable sanitario previo al uso de documentos nuevos o de documentos revisados y corregidos. Los documentos obsoletos deberán retirarse del personal que los utilice y deberán archivarse y conservarse.
20.1.12 Los manuales, guías, instructivos, documentos y registros apropiados y correctos deberán estar disponibles en todos los sitios donde las actividades esenciales lo requieran.
20.1.13 Los registros deberán ser completos y recobrables en un periodo de tiempo apropiado para las circunstancias y mantenerse protegidos contra destrucción o modificación accidental o no autorizada.
20.2 Expedientes del personal
Los expedientes del personal que realiza procesos críticos deberán contener la información siguiente:
20.2.1 Nombre, iniciales, código de identificación, firma y fecha de contratación;
20.2.2 La definición de la formación y la experiencia necesaria para un determinado cargo;
20.2.3 La duración del periodo de formación y de la experiencia;
20.2.4 Sus responsabilidades, actividades específicas asignadas y autorizadas;
20.2.5 En su caso, la capacitación otorgada por el establecimiento al inicio del cargo y durante el cargo y las evaluaciones de las mismas, y
20.2.6 Sus participaciones en programas de educación continua y de actualización en materia de medicina transfusional.
20.2.7 Evaluaciones periódicas del desempeño realizadas por el responsable sanitario o responsables de las áreas.
20.3 Documentación relativa a la disposición de sangre y productos sanguíneos.
20.3.1 Procedimientos normalizados de operación, guías e instructivos.
Los Bancos de Sangre, Centros de Colecta, Centros de Procesamiento, Centros de Calificación Biológica, Centros de Distribución de Sangre y Productos Sanguíneos y Servicios de Transfusión Hospitalario, en el ámbito de las funciones, deberán contar con procedimientos normalizados de operación, específicos y accesibles al personal que los emplee, en materia de lo siguiente:
20.3.1.1 Procedimientos normalizados de operación para el fomento de la donación voluntaria y altruista de sangre que deberá incluir lo siguiente:
20.3.1.1.1 Las actividades y procesos para efectuar para la promoción de la cultura de donación voluntaria y altruista de sangre;
20.3.1.1.2 Las actividades para asegurar que un donante voluntario y altruista pueda ser un donante regular o de repetición, y
20.3.1.1.3 La metodología para la planeación y programación de las campañas de donación voluntaria y altruista de sangre, en coordinación con los Centros Nacional y Estatales de Transfusión Sanguínea con el fin de fortalecer las redes nacionales y estatales de Bancos de Sangre, Centros de Colecta,
20.3.1.2 Procedimiento normalizado de operación para la atención y manejo de los donantes que deberá incluir entre otros:
20.3.1.2.1 La metodología para una atención digna y respetuosa, que permita ganar la empatía y confianza del donante a fin de que pueda obtenerse información veraz y que la evaluación médica resulte efectiva;
20.3.1.2.2 Los criterios para la aceptación, diferimiento o exclusión indefinida o permanente de los donantes conforme a lo señalado en el capítulo 7 denominado "Selección de donantes para uso terapéutico alogénico" de este proyecto de Norma;
20.3.1.2.3 La metodología para la aplicación del procedimiento de la autoexclusión, y
20.3.1.2.4 Las demás actividades o procedimientos que el establecimiento considere necesarios.
En el propio manual o de manera separada se contará con una lista actualizada de fármacos de uso común, con sus correspondientes periodos de diferimiento, incluyendo la tabla 5 de este proyecto de Norma.
20.3.1.3 Procedimientos normalizados de operación para la extracción de unidades de sangre, productos sanguíneos y muestras que incluya la metodología para la identificación adecuada de las unidades y muestras, así como para la detección, diagnóstico y manejo de incidentes y reacciones o efectos adversos a la donación.
20.3.1.4 Procedimientos normalizados de operación para el procesamiento, almacenaje, etiquetado, embalaje y traslado de unidades de sangre, productos sanguíneos o mezclas de éstos, reactivos y muestras, que incluyan las instrucciones a seguir en caso de falla en el suministro eléctrico o cualquier otra alteración en las condiciones del almacenamiento.
20.3.1.5 Procedimientos normalizados de operación para la realización de:
20.3.1.5.1 Las determinaciones analíticas aplicables a las muestras de las unidades de sangre o productos sanguíneos o bien, de las muestras de los receptores, y
20.3.1.5.2 El control de calidad de las unidades de sangre y productos sanguíneos.
20.3.1.6 Procedimientos normalizados de operación relativos a los criterios para la selección de unidades de acuerdo con el perfil inmunohematológico y la patología del receptor.
20.3.1.7 Instructivos para el uso y cuidados del equipamiento e instrumental crítico requerido para las actividades relativas a la disposición de sangre y productos sanguíneos que el establecimiento realiza, que deberán incluir información sobre el mantenimiento preventivo, los parámetros y frecuencia de revisión y el mantenimiento correctivo.
20.3.1.8 Procedimientos normalizados de operación de seguridad accesibles al personal expuesto a riesgos biológicos, físicos, mecánicos y químicos, que especifiquen las normas para manipulación, guarda y desecho de los materiales peligrosos. Estos procedimientos deberán contener, cuando menos, la información siguiente:
20.3.1.8.1 Clasificación de los agentes de riesgos sean: biológicos, químicos o físicos, indicando las medidas de prevención para cada uno de ellos;
20.3.1.8.2 Consideraciones generales de higiene, vestuario y protectores;
20.3.1.8.3 Instrucciones sobre la limpieza y desinfección del material y áreas de trabajo;
20.3.1.8.4 Instrucciones sobre la toma de muestras y su transportación, y
20.3.1.8.5 Conducta para seguir en caso de accidentes con riesgo biológico, químico o físicos.
20.3.1.9 Procedimiento normalizado de operación para el manejo de los residuos peligrosos biológico- infecciosos, que deberá contener, entre otros:
20.3.1.9.1 La clasificación y segregación de residuos;
20.3.1.9.2 La colecta, embalaje, manipulación y transporte de los residuos, y
20.3.1.9.3 Los demás que se requieran, en apego a la Norma referenciada en el inciso 2.20 de este proyecto de Norma.
20.3.1.10 Procedimiento normalizado de operación donde se describa de qué manera se coordina el servicio de sangre para sus actividades con otros servicios y establecimientos, debiendo incluir de los siguientes aquellos que correspondan a su caso: una coordinación efectiva entre:
20.3.1.10.1 Coordinación del servicio de sangre con los otros establecimientos que hacen disposición de sangre y productos sanguíneos con los que tenga convenios.
20.3.1.10.2 Coordinación del Banco de Sangre con los servicios de sangre que tenga bajo su responsabilidad de conformidad con el inciso 5.3 del presente proyecto de Norma.
20.3.1.10.3 Coordinación del servicio de sangre con Los establecimientos para la atención médica y, en su caso, los servicios clínicos del establecimiento de atención médica que le solicitan productos sanguíneos. (aplica para bancos de sangre intrahospitalarios y servicios de transfusión hospitalarios).
20.3.1.11 Procedimientos normalizados de operación o guías para el buen uso clínico de la sangre y productos sanguíneos accesibles al personal de salud de los bancos de sangre, servicios de transfusión y en los servicios clínicos de los establecimientos para la atención médica que participen en la indicación, aplicación y vigilancia de las transfusiones. (consúltese la guía referenciada en el inciso 22.10 de este proyecto de Norma). Estos documentos deberán incluir, entre otros, lo siguiente:
20.3.1.11.1 Indicaciones terapéuticas y contraindicaciones de los productos sanguíneos;
20.3.1.11.2 Metodología y vigilancia del acto transfusional;
20.3.1.11.3 Descripción y manejo de los incidentes, reacciones o efectos adversos a la transfusión, y
20.3.1.11.4 Los procedimientos analíticos, metodología para el registro y notificación al banco de sangre o servicio de transfusión de los incidentes, reacciones o efectos adversos a la transfusión.
20.3.2 Registros de ingresos y egresos de sangre y productos sanguíneos
Los Bancos de Sangre, Centros de Colecta, Centros de Procesamiento, Centros de Distribución de Sangre y Productos Sanguíneos y los Servicios de Transfusión Hospitalario, de acuerdo con las funciones que realizan, deberán contar con registros documentales sobre los ingresos y egresos de sangre y de sus componentes, que permitirán lo siguiente: Acceso a la identificación y localización de cada donante;
20.3.2.1.1 Una interrelación clara entre la fecha de donación, la identidad del donante y los componentes recolectados o preparados, por medio del número exclusivo para cada donación, y
20.3.2.1.2 La trazabilidad de las unidades desde su extracción hasta su uso terapéutico, destino final, o bien, suministro para la elaboración de hemoderivados, incluyendo los pasos intermedios.
20.3.2.2 La información concerniente a los ingresos y egresos de sangre y productos sanguíneos, podrá contenerse en un libro con hojas foliadas o en un conjunto de hojas agrupadas y numeradas en formato impreso o electrónico. Estos registros deberán contar con la aprobación y autorización de las autoridades sanitarias competentes.
20.3.2.3 Los registros documentales en el libro de registros de ingresos y egresos de sangre y productos sanguíneos o el conjunto de hojas foliadas, deberán reunir los requisitos siguientes:
20.3.2.3.1 Las hojas estarán foliadas (o numeradas);
20.3.2.3.2 La portada del libro o equivalente electrónico contendrá la información siguiente:
20.3.2.3.2.1 El nombre del propietario, la denominación del establecimiento, que incluirá el giro para el cual el establecimiento está autorizado;
20.3.2.3.2.2 El nombre del responsable sanitario;
20.3.2.3.2.3 El número de libro y la anotación del folio de la primera y última planas utilizables, y
20.3.2.3.2.4 Tratándose del libro de un servicio de sangre que se encuentre bajo la responsabilidad de un banco de sangre de conformidad con el inciso 5.3 del presente proyecto de Norma deberá contener la siguiente información en la portada:
20.3.2.3.2.5 nombre del propietario, la razón social y el nombre del responsable sanitario del banco de sangre del cual depende
20.3.2.3.2.6 modalidad del servicio de sangre del que se trate, nombre de su responsable sanitario y nombre del establecimiento para la atención médica donde se ubica.
20.3.2.3.2.7 En caso de cambio de responsable sanitario deberá registrase en la plana, renglón y fecha correspondiente del libro el nombre del nuevo titular, con las anotaciones que éste considere pertinentes a fin de deslindar responsabilidades. De manera similar se procederá en caso de cambio de encargado de un Centro de Colecta;
20.3.2.3.3 Los registros serán legibles e indelebles, o en su caso, con medidas de seguridad electrónica adecuadas, se mantendrán constantemente actualizados, sin raspaduras ni enmendaduras, y
20.3.2.3.4 De requerirse, las aclaraciones por errores o cambios de cualquier naturaleza se tacharán con una línea delgada de manera que queden legibles, los cambios o correcciones se harán entrerrenglonados o quedarán adecuadamente señalados y anotados en el propio libro o su equivalente electrónico.
20.3.2.4 Los registros en el libro o su equivalente electrónico, relativos a los ingresos y egresos de sangre y de sus componentes, que tenga un banco de sangre o un servicio de transfusión, incluirán, como mínimo, la información que indica la Tabla 36 de este proyecto de Norma.
Tabla 36
Información relativa a ingresos y egresos de sangre y productos sanguíneos de los bancos de sangre y servicios de transfusión
Ingresos |
Egresos |
a) Número progresivo para cada ingreso; b) Fecha de ingreso de las unidades de sangre o de sus componentes; c) Nombre del donante; d) El número exclusivo de identificación de la unidad, mismo que identificará también a los componentes fraccionados de la sangre total; e) En su caso, nombre del establecimiento o del Centro de Colecta de procedencia de cada unidad; f) El uso terapéutico que se le pretende dar, entre las siguientes: alogénica, autóloga o singénica; g) El señalamiento del tipo de donación: - Voluntaria y altruista; - Familiar o de reposición; |
a) Fecha y hora de egreso del componente sanguíneo; b) Nombre del establecimiento al que se suministró la unidad, o bien, cuando proceda nombre del receptor, su número exclusivo de expediente o registro, fecha de nacimiento, en su caso, el domicilio donde se llevará a cabo la transfusión; c) En su caso, el motivo del destino final; d) Clasificación de grupo AB0 y Rh (D) del receptor; e) Nombre del médico que indica la transfusión; f) Volumen egresado cuando se trate de unidades para uso pediátrico; g) Cualquier eventualidad que requiera ser consignada, y |
- Designada; - Dirigida; - Regular, o - De repetición. h) El método de extracción: (extracción habitual de sangre total o de componentes mediante aféresis); i) El señalamiento del contenido de la unidad; j) Clasificación del grupo AB0 y Rh (D); k) Los bancos de sangre registrarán los resultados de las pruebas obligatorias para la detección de enfermedades transmisibles por transfusión, así como los resultados de otras pruebas que se hubiesen practicado; l) Procesamientos efectuados a las unidades, tales como: lavado, leucodepleción mediante filtrado, irradiación, inactivación u otros; m) Fecha de caducidad de la unidad; n) Cualquier eventualidad que requiera consignarse, y o) Las demás que el establecimiento considere necesarias. |
h) Las demás que el establecimiento considere necesarias. |
20.3.2.5 Los registros en el libro sobre los ingresos y egresos de sangre y de sus componentes que tenga un Centro de Colecta, deberá contener cuando menos la información que indica la Tabla 37 de este proyecto de Norma.
Tabla 37
Información relativa a ingresos y egresos de sangre y productos sanguíneos de los Centro de Colecta
Ingresos |
Egresos |
a) El número progresivo exclusivo para la unidad de sangre extraída; b) Nombre del donante; c) Fecha y hora de extracción de las unidades de sangre; d) El uso terapéutico que se le pretende dar, entre las siguientes: alogénica, autóloga o singénica; e) El señalamiento del tipo de donación: - Voluntaria y altruista; - Familiar o de reposición; - Designada; - Dirigida; - Regular, y - De repetición f) Las demás que el establecimiento considere necesarias. |
a) Fecha y hora de envío al banco de sangre del cual depende el Centro de Colecta; b) En su caso, motivo del destino final; c) Cualquier eventualidad que requiera ser consignada, y d) Las demás que el establecimiento considere necesarias. |
20.3.3 Cartas de consentimiento informado.
20.3.3.1 La carta de consentimiento informado en la que un donante consiente la donación de sangre o productos sanguíneos para uso alogénico deberá obtenerse en cada donación y contendrá lo siguiente:
20.3.3.1.1 Nombre del banco de sangre o Centro de Colecta, ubicación y, en su caso, la institución a la que pertenece;
20.3.3.1.2 Nombre o título del documento;
20.3.3.1.3 Nombre, sexo, identidad de género, edad, domicilio, ocupación y estado civil del donante;
20.3.3.1.4 El tipo de donación: voluntaria y altruista, familiar o de reposición, designada, dirigida, o bien, regular o de repetición;
20.3.3.1.5 Objetivos del acto de disposición, beneficios y posibles riesgos para el receptor;
20.3.3.1.6 Información sobre los procedimientos que vayan a efectuarse, que incluya:
20.3.3.1.6.1 Método de colecta;
20.3.3.1.6.2 Los volúmenes de sangre o productos sanguíneos que pretendan obtenerse;
20.3.3.1.6.3 Las posibles reacciones o efectos adversos que pudieran presentarse;
20.3.3.1.6.4 En su caso, las soluciones o fármacos que fuesen a usarse y su propósito, incluyendo la identificación de aquellos que estén en el proceso de evaluación experimental, así como la información sobre toxicidad, efectos secundarios, dosis, tiempo y costo del tratamiento, y
20.3.3.1.6.5 Si los hubiese, los procedimientos alternativos;
20.3.3.1.7 Recomendaciones para después de la donación incluyendo, en su caso, la necesidad de esperar un intervalo desde la donación hasta la vuelta a una actividad profesional o de afición que conlleve riesgos.
20.3.3.1.8 Información sobre las determinaciones analíticas que se efectuarán, especificando los análisis para la detección de enfermedades transmisibles por transfusión y en la eventualidad que resultasen anormales, que se dará destino final a la sangre o componentes recolectados y sobre la posibilidad de futuras citas para la toma de nuevas muestras;
20.3.3.1.9 Tratándose de donantes designados, además de lo anterior contendrá:
20.3.3.1.9.1 El nombre, sexo, identidad de género, edad, domicilio, ocupación y estado civil del receptor, así como los riesgos y beneficios esperados para éste, y
20.3.3.1.9.2 La probabilidad de una segunda donación o secuencia de donaciones para el mismo receptor;
20.3.3.1.10 Las declaraciones siguientes:
20.3.3.1.10.1 Que recibió información a su satisfacción sobre los riesgos y consecuencias de la donación;
20.3.3.1.10.2 Que leyó y entendió la información y el material educativo que le fue proporcionado;
20.3.3.1.10.3 Que se le brindó la oportunidad de hacer preguntas y que éstas fueron contestadas satisfactoriamente por un profesional capacitado;
20.3.3.1.10.4 Que la información aportada por el donante es, a su juicio, veraz y sincera;
20.3.3.1.10.5 Que está de acuerdo en que se realicen las pruebas de detección de enfermedades transmisibles por transfusión y que, de resultar anormales, será notificado personalmente o a quien designe para ello, a través de una carta poder firmada ante dos testigos, y
20.3.3.1.10.6 Que, por propia voluntad y título gratuito, consiente la donación de su sangre o de productos sanguíneos;
20.3.3.1.11 Firma o huella dactilar del donante, y
20.3.3.1.12 Lugar y fecha en que se emite.
20.3.3.2 La carta de consentimiento informado para efectuar procedimientos de transfusión autóloga deberá incluir la información siguiente:
20.3.3.2.1 Nombre, ubicación y, en su caso, nombre de la institución a la que pertenece el banco de sangre, servicio de transfusión o establecimiento de atención médica que efectuará el procedimiento;
20.3.3.2.2 Nombre o título del documento;
20.3.3.2.3 Nombre, sexo, identidad de género, edad, domicilio, ocupación y estado civil del paciente;
20.3.3.2.4 El diagnóstico de probabilidad o certeza;
20.3.3.2.5 El o los procedimientos de transfusión autóloga que se efectuarán;
20.3.3.2.6 Los objetivos del procedimiento, beneficios y posibles riesgos, reacciones o efectos adversos que pudieran presentarse;
20.3.3.2.7 Los volúmenes de sangre o productos sanguíneos que se pretendan extraer;
20.3.3.2.8 En su caso, las soluciones o fármacos que fuesen a usarse y su propósito, incluyendo la identificación de aquellos que estén en el proceso de evaluación experimental, así como la información sobre toxicidad, efectos secundarios, dosis, tiempo y costo del procedimiento;
20.3.3.2.9 Información sobre la posibilidad de que se requiera el uso de sangre o productos sanguíneos alogénicos;
20.3.3.2.10 Tratándose de procedimientos de transfusión autóloga mediante depósito previo, la carta de consentimiento incluirá además lo siguiente:
20.3.3.2.10.1 Información sobre las determinaciones analíticas que se efectuarán, especificando los análisis para la detección de enfermedades infecciosas transmisibles por transfusión y en la eventualidad que resultasen anormales, la necesidad de excluirlo del procedimiento, del destino final que se dará a la sangre o productos sanguíneos colectados y sobre la posibilidad de futuras citas para la obtención de muestras sanguíneas adicionales, y
20.3.3.2.10.2 Las recomendaciones para después de cada extracción.
20.3.3.2.11 Las declaraciones siguientes:
20.3.3.2.11.1 Que recibió información a su satisfacción sobre los riesgos y beneficios del procedimiento de transfusión autóloga;
20.3.3.2.11.2 Que leyó y entendió la información y el material educativo que le fue proporcionado;
20.3.3.2.11.3 Que se le brindó la oportunidad de hacer preguntas y que éstas fueron contestadas satisfactoriamente por un profesional capacitado;
20.3.3.2.11.4 En procedimientos de autotransfusión por depósito previo, que consiente la realización de las pruebas de detección de enfermedades infecciosas transmisibles por transfusión y que, de resultar anormales, será excluido del procedimiento y del destino final de las unidades de sangre o componentes recolectados, y
20.3.3.2.11.5 Que por propia voluntad y con pleno conocimiento de causa consiente el procedimiento de que se trate y que autoriza al personal de salud para la atención de contingencias derivadas del acto consentido, atendiendo al principio de autoridad prescriptiva;
20.3.3.2.12 En caso de pacientes menores o en estado de interdicción de otorgar el consentimiento incluirá lo siguiente:
20.3.3.2.12.1 El nombre, sexo, identidad de género, edad, domicilio, ocupación y estado civil de quien otorga el consentimiento, y
20.3.3.2.12.2 El vínculo con el paciente que le permite ejercer tal derecho y la causa por la que lo ejerce;
20.3.3.2.13 Firma o huella dactilar del paciente o, en su caso, del otorgante del consentimiento, y
20.3.3.2.14 Lugar y fecha en que se emite.
La carta de consentimiento informado para procedimientos de hemodilución aguda preoperatorio o recuperación sanguínea perioperatoria, podrá ser en formatos independientes o estar incluidos en el documento en el que el paciente consiente el procedimiento quirúrgico de que se trate.
20.3.3.3 La carta de consentimiento informado en el que un receptor expresa su consentimiento para recibir una transfusión deberá contener:
20.3.3.3.1 Nombre del establecimiento, ubicación y, en su caso, nombre de la institución a la que pertenece;
20.3.3.3.2 Nombre o título del documento;
20.3.3.3.3 Nombre, sexo, identidad de género, edad, domicilio, ocupación y estado civil del paciente;
20.3.3.3.4 Información sobre el o los productos sanguíneos que serán transfundidos, los objetivos, beneficios y posibles riesgos y, en su caso, los procedimientos alternativos que hubiese;
20.3.3.3.5 El diagnóstico de probabilidad o certeza;
20.3.3.3.6 Las declaraciones siguientes:
20.3.3.3.6.1 Que recibió información a su satisfacción sobre los riesgos y consecuencias de la transfusión;
20.3.3.3.6.2 Que el receptor leyó y entendió la información y el material educativo proporcionado;
20.3.3.3.6.3 Que se le brindó la oportunidad de hacer preguntas y que éstas fueron contestadas satisfactoriamente por un profesional capacitado;
20.3.3.3.6.4 Que por propia voluntad y con pleno conocimiento de causa consiente la transfusión de que se trate y que autoriza al personal de salud para la atención de contingencias derivadas del acto consentido, atendiendo al principio de autoridad prescriptiva;
20.3.3.3.7 Firma o huella dactilar del paciente, y
20.3.3.3.8 Lugar y fecha en que se emite.
En casos de urgencia podrá omitirse el consentimiento para recibir una transfusión y se estará a lo dispuesto a lo señalo por el artículo 81 del Reglamento de la Ley General de Salud en materia de Atención Médica.
20.3.3.4 La carta de consentimiento informado para transfundir a menores de edad o en estado de interdicción, en la que una persona capaz de otorgar el consentimiento consiente que la transfusión se lleve a cabo, deberá contener la información siguiente:
20.3.3.4.1 Nombre del establecimiento, ubicación y, en su caso, de la institución a la que pertenece;
20.3.3.4.2 Nombre o título del documento;
20.3.3.4.3 Nombre, sexo, identidad de género, edad, domicilio, ocupación y estado civil del paciente;
20.3.3.4.4 El diagnóstico de probabilidad o certeza;
20.3.3.4.5 Nombre, sexo, identidad de género, edad, domicilio, ocupación y estado civil de quien otorga el consentimiento;
20.3.3.4.6 El vínculo con el paciente que le permite ejercer tal derecho y la causa por la que lo ejerce;
20.3.3.4.7 Información sobre el o los productos sanguíneos que serán transfundidos, los objetivos, beneficios y posibles riesgos y, en su caso, los procedimientos alternativos que hubiese;
20.3.3.4.8 Las declaraciones siguientes:
20.3.3.4.9 Que recibió información a su satisfacción sobre los riesgos y consecuencias de la transfusión, que se le brindó la oportunidad de hacer preguntas y que éstas fueron contestadas satisfactoriamente por un profesional capacitado;
20.3.3.4.10 Que el otorgante del consentimiento leyó y entendió la información y el material educativo proporcionado, y
20.3.3.4.11 Que por propia voluntad y con pleno conocimiento de causa consiente la transfusión de que se trate y que autoriza al personal de salud para la atención de contingencias derivadas del acto consentido, atendiendo al principio de autoridad prescriptiva;
20.3.3.4.12 Firma o huella dactilar del otorgante, y
20.3.3.4.13 Lugar y fecha en que se emite.
En casos de urgencia para transfundir a un menor o un incapaz podrá omitirse el consentimiento para recibir la y se estará a lo dispuesto a lo señalo por el artículo 81 del Reglamento de la Ley General de Salud en materia de Atención Médica.
20.3.4 Otros documentos, formatos y registros.
20.3.4.1 Historia clínica del donante.
Los bancos de sangre y los Centro de Colecta deberán practicar a todos los candidatos a donar una historia clínica, en un formato aprobado para ello por la Secretaría, que tendrá carácter confidencial y contendrá, como mínimo, la información siguiente:
20.3.4.1.1 Fecha de su elaboración;
20.3.4.1.2 El número exclusivo de la unidad de sangre o componente sanguíneo que se obtuvo;
20.3.4.1.3 Los datos del donante que permitan su identificación y localización, que incluyan:
20.3.4.1.3.1 Nombre y firma o huella dactilar del donante;
20.3.4.1.3.2 Documento con el que se identifica, dependencia que lo emite y el número del documento;
20.3.4.1.3.3 Edad y fecha de nacimiento;
20.3.4.1.3.4 Sexo e identidad de género;
20.3.4.1.3.5 Ocupación, y
20.3.4.1.3.6 Domicilio, teléfono y, en su caso, correo electrónico;
20.3.4.1.3.7 Uso terapéutico que se le pretenda dar a las unidades extraídas, entre las posibilidades siguientes: donación para uso alogénico, singénico o autólogo;
20.3.4.1.4 El señalamiento del tipo de donación:
20.3.4.1.4.1 Voluntaria y altruista;
20.3.4.1.4.2 Familiar o de reposición;
20.3.4.1.4.3 Designada;
20.3.4.1.4.4 Dirigida;
20.3.4.1.4.5 Regular, o
20.3.4.1.4.6 De repetición;
20.3.4.1.5 Método de extracción que podrá ser donación de sangre total o de productos sanguíneos mediante métodos de aféresis;
20.3.4.1.6 Tratándose de donantes regulares o de repetición, la cantidad, tipo de donación y fecha de la última y de ser posible las fechas de las donaciones anteriores. En caso de donantes de repetición, además de lo anterior, los datos del Banco de Sangre o Centro de Colecta donde donó previamente;
20.3.4.1.7 Los datos relevantes que permitan identificar riesgos potenciales para la salud del donante o de los receptores, de conformidad con los criterios de aceptabilidad o exclusión que establece el capítulo 7 denominado "Selección de donantes para uso terapéutico alogénico" de este proyecto de Norma;
20.3.4.1.8 Descripción detallada de todas las actividades o procedimientos realizados, productos farmacéuticos empleados y resultados obtenidos, que incluya, como mínimo, lo siguiente:
20.3.4.1.8.1 El volumen de sangre o componentes obtenidos;
20.3.4.1.8.2 La selección de anticoagulantes, soluciones y medicamentos utilizados, así como su dosificación;
20.3.4.1.8.3 Los resultados del procedimiento, incluyendo, en su caso, la descripción de complicaciones, reacciones o eventos adversos que se hubiesen presentado y su manejo, y
20.3.4.1.8.4 En donaciones para transfusión autóloga mediante procedimientos de depósito previo, la frecuencia de las recolecciones señaladas por fecha y el volumen obtenido en cada sesión;
20.3.4.1.9 En la propia historia o anexa a ella, estarán los resultados de las determinaciones analíticas efectuadas;
20.3.4.1.10 Razones por las cuales se hubiera excluido o diferido al donante o, en su caso, motivo por el cual se hubiera dado destino final a su sangre o productos sanguíneos, y
20.3.4.1.11 Nombre y firma del médico que hizo la evaluación del donante y del personal de salud que realizó los procedimientos de extracción.
20.3.4.2 Autoexclusión confidencial.
El procedimiento de autoexclusión confidencial se llevará a cabo conforme a lo señalado en los subincisos incluidos en el inciso 7.9 de este proyecto de Norma.
Los documentos empleados para la autoexclusión en los que un donante manifiesta confidencialmente, si considera su sangre o productos sanguíneos aptos o no para uso transfusional, deberán reunir los requisitos y contenido que señala la Tabla 38 de este proyecto de Norma.
Tabla 38
Documentos aplicables para el procedimiento de autoexclusión confidencial
Requisitos del material informativo y educativo que se debe proporcionar al donante |
Contenido del formato o impreso de respuesta de la autoexclusión del donante |
a) La información se otorgará por escrito, expresada de manera clara y entendible. Es recomendable que la información se presente de manera audiovisual o, en su defecto, con el uso de rotafolio; b) Tenderá a motivar y sensibilizar al donante para que la información que le es requerida la otorgue con precisión veracidad, y c) El material informativo y educativo deberá incluir: |
a) El formato o impreso se identificará exclusivamente con el número exclusivo asignado a la unidad de sangre o componente sanguíneo extraído; b) Ofrecerá dos opciones de respuesta para que el donante señale en una de ellas, con una "X"; si considera apta o no su sangre o productos sanguíneos para uso terapéutico, y |
- El objetivo de la autoexclusión e información de la existencia de periodos de ventana de enfermedades infecciosas transmisibles por transfusión, durante los cuales debe evitarse la donación; |
c) Tendrá la información necesaria, conforme al mecanismo adoptado por el establecimiento, para asegurar que el donante lo responda y lo haga llegar al personal asignado del banco de sangre o Centro de Colecta. |
- Información que lo habilite a identificar las prácticas sexuales y cualquier otro evento de riesgo que pudiese haberse expuesto para adquirir una infección transmisible por transfusión, e |
|
- Información sobre la manera de proceder con el impreso donde manifestará si considera apta o no su sangre o productos sanguíneos para uso terapéutico. |
20.3.4.3 Expedientes de los donantes
El expediente de los donantes deberá contener los documentos siguientes:
20.3.4.3.1 La carta de consentimiento informado otorgado que incluirá: el nombre y firma o huella dactilar del donante y el número exclusivo de la unidad recolectada;
20.3.4.3.2 La historia clínica a que se refiere el inciso 20.3.4.1 de este proyecto de Norma;
20.3.4.3.3 En donaciones para uso alogénico o singénico, el impreso en que el donante manifiesta si considera adecuada su sangre o productos sanguíneos para uso transfusional, y
20.3.4.3.4 Copia del documento otorgado a un donante donde se le proporciona orientación para que obtenga atención médica, en caso de haberse detectado o sospechado la presencia de cualquier enfermedad a través de la evaluación médica o de las determinaciones analíticas efectuadas.
20.3.4.3.5 Formato para el envío de unidades de sangre de un Centro de Colecta al Banco de Sangre del cual depende.
20.3.4.3.6 Los Centros de Colecta deberán tener un documento en el que conste el envío de unidades de sangre al banco del cual dependen, el original acompañará la remesa y conservarán la copia. El documento estará escrito con letra legible y contendrá, como mínimo, la información siguiente:
20.3.4.3.7 Nombre del Centro de Colecta, su ubicación y, en su caso, de la institución a la que pertenece;
20.3.4.3.8 Datos de identificación del banco de sangre del cual depende;
20.3.4.3.9 Número progresivo exclusivo para la unidad de sangre y nombre del donante;
20.3.4.3.10 Fecha y hora de salida de las unidades del Centro de Colecta y de su llegada al Banco de Sangre;
20.3.4.3.11 Nombre y firma de quien preparó las unidades para su envío, así como, de quien recibe, y
20.3.4.3.12 Observaciones al momento de la recepción, donde se anotará cualquier irregularidad relativa al embalaje, eventualidades durante el traslado, la identificación de las unidades, estado físico, trazabilidad de la temperatura o, en su caso, estimación de ésta, o cualquier otra eventualidad que amerite registrarse.
20.3.4.4 Registro de las determinaciones analíticas.
El personal del laboratorio de un Banco de Sangre, Servicio de Transfusión hospitalarios y Centro de Procesamiento deberá revisar, registrar y mantener archivados los resultados de las determinaciones analíticas de cada donante o cada receptor estudiado. Los registros incluirán, cuando menos, la información siguiente:
20.3.4.4.1 Nombre del donante y el número exclusivo asignado a la unidad de sangre o productos sanguíneos;
20.3.4.4.2 Nombre completo del receptor y, en su caso, fecha de nacimiento, número de expediente y número de cama o habitación;
20.3.4.4.3 Fecha y hora de ejecución, metodología empleada y resultados de las pruebas de detección de agentes transmisibles por transfusión. De emplearse técnicas inmunoenzimáticas, deberá conservarse el registro o impresión original de los resultados de las pruebas;
20.3.4.4.4 Fecha y hora de ejecución, metodología empleada y resultados de las pruebas de inmunohematología, incluyendo la compatibilidad entre un donante y un receptor;
20.3.4.4.5 Resultados de las pruebas efectuadas y su interpretación;
20.3.4.4.6 Nombre de los reactivos utilizados, la identificación del fabricante, distribuidor o ambos, el número de lote las fechas de recepción y de caducidad;
20.3.4.4.7 Nombre y firma de la persona autorizada que realizó el estudio, y
20.3.4.4.8 En su caso, nombre y firma del personal que revisó y reportó el estudio.
20.3.4.5 Registros de control de calidad.
20.3.4.5.1 Los Servicios de Sangre, de acuerdo con las actividades que desempeñan, deben llevar los registros siguientes:
20.3.4.5.1.1 Control de calidad que hagan a sus reactivos, equipos y técnicas, de conformidad con lo que establece este proyecto de Norma, las Normas Oficiales Mexicanas aplicables y las instrucciones proporcionadas por el fabricante, y
20.3.4.5.2 De los procedimientos referentes a las pautas para asegurar la calibración y mantenimiento del equipamiento o instrumental empleado en actividades críticas, que incluirá, entre otros, la información siguiente:
20.3.4.5.2.1 Identificación del equipo, modelo y número de serie u otra identificación única;
20.3.4.5.2.2 Nombre del fabricante o distribuidor;
20.3.4.5.2.3 La condición de uso a la recepción, es decir, si es nuevo, utilizado o reacondicionado;
20.3.4.5.2.4 Fecha de instalación y fecha de entrada en funcionamiento;
20.3.4.5.2.5 Cada equipo dispondrá de un registro individual donde conste su mantenimiento, si éste fue externo o interno o bien, preventivo o correctivo, indicando con los parámetros o aspectos controlados, fecha y resultado y el responsable de su realización, y
20.3.4.5.2.6 Un plan o calendario con las calibraciones y verificaciones que deberán efectuarse en los equipos de que se trate. Cada equipo dispondrá de un registro individual donde conste su mantenimiento, si éste fue externo o interno o bien, preventivo o correctivo, indicando con los parámetros o aspectos controlados, fecha y resultado y el responsable de su realización, y
20.3.4.5.2.7 Un plan o calendario con las calibraciones y verificaciones que deberán efectuarse en los equipos de que se trate.
20.3.4.6 Solicitud de productos sanguíneos
La solicitud de unidades de sangre, productos sanguíneos o mezclas de éstos, deberá contener como mínimo la información siguiente:
20.3.4.6.1 Datos de identificación del establecimiento, servicio clínico o establecimiento de atención médica que hace la solicitud, que incluya razón social, domicilio y teléfono;
20.3.4.6.2 Datos de identificación y localización del receptor:
20.3.4.6.2.1 Nombre y edad, cuando sea posible;
20.3.4.6.2.2 Sexo e identidad de género, y
20.3.4.6.2.3 Tratándose de pacientes hospitalizados, nombre completo, fecha de nacimiento, el número exclusivo de expediente o registro, el número de cama o habitación y, en su caso, el nombre del servicio;
20.3.4.6.3 De conocerse, clasificación de grupo sanguíneo AB0 y Rh (D), antecedentes transfusionales, gestacionales, de inmunización materno fetal o de reacciones o efectos adversos en transfusiones previas;
20.3.4.6.4 Diagnóstico de certeza o de probabilidad;
20.3.4.6.5 Motivo de la indicación transfusional y las cifras de laboratorio del parámetro hematológico que se busca mejorar;
20.3.4.6.6 El señalamiento de lo solicitado, unidad o mezcla de unidades, cantidad y, en su caso, volumen o características específicas requeridas;
20.3.4.6.7 En su caso, el señalamiento de que se trata de transfusión para uso autólogo;
20.3.4.6.8 Cuando proceda, fecha y hora en que se realizará la transfusión;
20.3.4.6.9 En caso de solicitudes urgentes, el motivo del apremio, así como si el requerimiento es inmediato o si puede diferirse 30 minutos;
20.3.4.6.10 Fecha y hora de la solicitud, y
20.3.4.6.11 Nombre del médico que indica la transfusión y, en su caso, nombre y firma del solicitante, así como, el teléfono fijo o móvil de cualquiera de las personas a que se refiere este inciso, para que, en caso necesario, sea posible su localización.
20.3.4.7 Marbete anexo a los productos sanguíneos.
Toda unidad de sangre, de productos sanguíneos o mezclas de éstos egresadas por un banco de sangre o un servicio de transfusión deberá acompañarse de un marbete que contendrá la información siguiente:
20.3.4.7.1 Datos de identificación del banco de sangre o servicio de transfusión que realiza el egreso;
20.3.4.7.2 Datos de identificación y localización del receptor:
20.3.4.7.2.1 Nombre y edad del receptor, cuando sea posible;
20.3.4.7.2.2 Sexo e identidad de género del receptor;
20.3.4.7.3 Tratándose de pacientes hospitalizados, nombre completo, fecha de nacimiento, el número exclusivo de expediente o registro, el número de cama o habitación y, en su caso, el nombre del servicio, y diagnóstico de probabilidad o certeza;
20.3.4.7.4 Los datos relativos a la fecha y hora de inicio de la transfusión, su vigilancia y reporte de los eventos, reacciones o efectos adversos a la transfusión;
20.3.4.7.5 Las instrucciones relativas a lo que se indica a continuación:
20.3.4.7.5.1 Sobre la conservación de las unidades hasta antes de su aplicación terapéutica, que especifiquen los tiempos máximos en que las unidades puedan estar fuera de los rangos de temperatura adecuados para evitar daño en su viabilidad y otros riesgos;
20.3.4.7.5.2 Sobre el acto transfusional, que incluyan: tiempos recomendados para la transfusión según el componente de que se trate y el uso de filtros para la retención de coágulos;
20.3.4.7.5.3 En su caso, instrucciones para el descongelamiento de los plasmas o crioprecipitados, señalando la prohibición de su recongelamiento y las demás que se requieran de acuerdo con el componente de que se trate, y
20.3.4.7.5.4 Metodología para la toma de muestras en caso de presentarse reacciones o eventos adversos a la transfusión, y
20.3.4.7.6 El señalamiento de que el marbete debidamente llenado, deberá ser devuelto al banco de sangre o al servicio de transfusión que suministró la unidad.
20.3.4.8 Registro del suministro y recepción de productos sanguíneos
Los servicios de sangre deberán contar con un formato para el registro de las unidades de sangre, productos sanguíneos o muestras de éstos, que envíen o reciban de establecimientos similares. Estos registros deberán contener la información siguiente:
20.3.4.8.1 Datos de identificación del servicio de sangre que hace el envío y, en su caso, número único del egreso;
20.3.4.8.2 Datos de identificación del establecimiento al que se envían las unidades o mezclas;
20.3.4.8.3 Cantidad y nombre de las unidades o mezclas, con su correspondiente identificación numérica o alfanumérica exclusiva para cada unidad o mezcla enviada y, en su caso, de las muestras sanguíneas y los demás documentos y registros que la acompañen;
20.3.4.8.4 Grupo sanguíneo AB0 y Rh (D) de las unidades;
20.3.4.8.5 Fecha y hora de extracción, así como de la caducidad de las unidades o mezclas; Condiciones en que se efectúa el embalaje;
20.3.4.8.6 Fecha y hora del envío;
20.3.4.8.7 Nombre y firma del personal del banco de sangre o servicio de transfusión que entrega el o los productos sanguíneos;
20.3.4.8.8 Fecha, hora y condiciones en que se reciben los productos sanguíneos;
20.3.4.8.9 Nombre y firma de quien recibe, y
20.3.4.8.10 Observaciones al momento de la recepción, donde se anotará cualquier irregularidad en las unidades o mezclas en lo relativo a su identificación, estado físico, estimación de su temperatura, contenido de aire, u otras eventualidades.
20.3.4.9 Registros de las transfusiones
Los bancos de sangre, los servicios de transfusión y los servicios clínicos de los establecimientos de atención médica que apliquen transfusiones, deberán registrar en un libro, el número de productos sanguíneos transfundidos, Estos registros deberán contener la información siguiente:
20.3.4.9.1 Datos de identificación del establecimiento que aplica la transfusión;
20.3.4.9.2 Tratándose de establecimientos de atención médica el nombre completo y fecha de nacimiento del paciente, número de cama o habitación donde se encuentra el paciente y cuando resulte aplicable el nombre del servicio clínico;
20.3.4.9.3 Nombre del banco de sangre o servicio de transfusión que suministró el o los productos sanguíneos. Datos de identificación del receptor:
20.3.4.9.3.1 Nombre y edad, cuando sea posible;
20.3.4.9.3.2 Sexo e identidad de género, y
20.3.4.9.3.3 Número exclusivo de expediente o registro;
20.3.4.9.4 El grupo AB0 y Rh (D) del paciente que recibirá la transfusión, salvo casos de transfusión de urgencia en la que se desconozca el grupo sanguíneo;
20.3.4.9.5 Diagnóstico de probabilidad o certeza;
20.3.4.9.6 Nombre y número exclusivo de las unidades o mezcla de productos sanguíneos, el grupo AB0 y Rh (D) de las unidades;
20.3.4.9.7 En su caso, registrará cualquier anomalía detectada en el envío, misma que la notificará al banco de sangre o servicio de transfusión;
20.3.4.9.8 Fecha y hora de la recepción e inicio de la transfusión;
20.3.4.9.9 Los datos relativos a la vigilancia del acto transfusional y el volumen del componente transfundido;
20.3.4.9.10 En su caso, información adecuada y suficiente sobre cualquier incidente, reacción o efecto adverso que se presente o detecte durante o después de la transfusión, que incluya el tiempo transcurrido entre el inicio de la transfusión y la aparición de los síntomas o signos de la reacción o efecto adverso;
20.3.4.9.11 El manejo de los efectos o reacciones adversas y sus resultados;
20.3.4.9.12 En su caso, el reporte de las determinaciones analíticas, efectuadas por el banco de sangre o servicio de transfusión que suministró los productos sanguíneos, tendientes a investigar las causas de la reacción o efecto adverso, y
20.3.4.9.13 Fecha, nombre y firma del médico que prestó la atención médica, así como el nombre del personal del banco de sangre o servicio de transfusión que intervino en las determinaciones analíticas pre y postransfusionales.
Además de los registros a que hace referencia este apartado, los establecimientos que apliquen transfusiones establecerán la prevalencia de eventos y reacciones adversas por unidades transfundidas.
20.3.4.10 Registros de las transfusiones en el expediente clínico del receptor
Los registros de las transfusiones aplicadas que se hagan en el expediente clínico del receptor deberán contener, como mínimo, la información siguiente:
20.3.4.10.1 Cantidad de unidades o mezclas de éstas;
20.3.4.10.2 El número exclusivo de identificación de cada unidad o mezcla de productos sanguíneos;
20.3.4.10.3 Fecha, hora de inicio y término de la transfusión;
20.3.4.10.4 En transfusión de sangre, concentrados de eritrocitos y plasma, el control de los signos vitales y el estado general del paciente, antes, durante y después de la transfusión;
20.3.4.10.5 En transfusión de concentrados de plaquetas y crioprecipitados, el control de signos vitales y el estado general del paciente antes y después de la transfusión;
20.3.4.10.6 En caso de reacciones adversas a la transfusión indicar su tipo y manejo, así como los procedimientos para efectos de la investigación correspondiente, y
20.3.4.10.7 Nombre y firma del médico que indicó la transfusión, así como del personal de salud encargado de la aplicación, vigilancia y control de la transfusión.
20.3.5 Tiempo de conservación de documentos y registros. Los servicios de sangre que hacen disposición de sangre y productos sanguíneos, deberán conservar los documentos a que hace referencia este capítulo, por los lapsos que se indican en la Tabla 39 de este proyecto de Norma.
Tabla 39
Tiempo de conservación de documentos y registros
Documento o registro |
Tiempo mínimo |
|
20.3.5.1 |
Revisiones del sistema de gestión de calidad, incluyendo, entre otros, organigrama y competencias del personal. |
Tres años |
20.3.5.2 |
Políticas, procesos, procedimientos, manuales, guías e instructivos remplazados y resultados de la validación de nuevos procesos. |
Tres años |
20.3.5.3 |
Expedientes del personal que realiza procesos críticos, que incluyan: a) En su caso, claves de acceso, y b) Información relativa a calificaciones, entrenamiento, aptitudes, educación continuada y otros. |
Cinco años |
20.3.5.4 |
Resultados de auditorías al sistema de gestión de calidad. |
Tres años |
20.3.5.5 |
Visitas de verificación sanitaria o visitas de supervisión y orientación por parte de los Centros Nacional o Estatales de la transfusión Sanguínea. |
Tres años. |
20.3.5.6 |
Registro de incidencias o no conformidades y de las acciones correctivas. |
Tres años |
20.3.5.7 |
El libro o equivalente para el registro de ingresos y egresos de sangre y sus componentes para la trazabilidad de cada unidad y sus fracciones. |
A partir del momento de su cancelación: - Cinco años en archivo activo, y - Cinco años en archivo muerto |
20.3.5.8 |
Expedientes de los donantes, incluyendo: - Historial clínico y método de extracción; - Consentimiento informado para donación alogénica o para someterse a un procedimiento de transfusión autóloga; - Resultados de la autoexclusión, y - Resultados de las determinaciones analíticas. |
A partir de la última donación: - Cinco años en archivo activo, y - Cinco años en archivo muerto |
20.3.5.9 |
Análisis de los motivos de exclusión de los donantes y la prevalencia de los mismos. |
Tres años |
20.3.5.10 |
Registro de donantes que han sido rechazados permanentemente. |
Cinco años |
20.3.5.11 |
Registro de las reacciones adversas a la donación. |
Tres años |
20.3.5.12 |
Registro de la información relativa al procesamiento de unidades. |
Tres años |
20.4 Resultados de las pruebas para detección de enfermedades infecciosas transmisibles por transfusión en los donantes y unidades: |
- Diez años en archivo activo, y - Cinco años en archivo muerto |
|
20.4.1 |
Resultados de las pruebas de inmunohematología en las unidades y los receptores, incluyendo las de compatibilidad sanguínea y, en su caso, de las pruebas realizadas para investigar las reacciones o efectos adversos a la transfusión. |
Cinco años |
20.4.2 |
Inventario del equipo empleado en procesos críticos, registros de pruebas, registros de temperaturas de almacenamiento, mantenimiento y verificaciones. |
Tres años |
Documento o registro |
Tiempo mínimo |
|
20.4.3 |
Pruebas de control calidad de los reactivos empleados en procesos críticos. |
Cinco años |
20.4.4 |
Pruebas de control de calidad efectuadas en los productos sanguíneos. |
Tres años |
20.4.5 |
Registros de la notificación al donante y al receptor de anomalías en las determinaciones analíticas. |
Tres años |
20.4.6 |
Registro de las actividades realizadas para efectos del suministro de las unidades. |
Tres años |
20.4.7 |
Registro de las transfusiones aplicadas por los bancos de sangre, los servicios de transfusión hospitalarios y los servicios clínicos. |
Tres años |
20.4.8 |
Consentimiento informado para recibir una transfusión y registros clínicos relativos al acto transfusional. |
Conforme con la Norma referenciada en el inciso 2.124 de este proyecto de Norma |
20.4.9 |
Investigación analítica de presuntas infecciones imputables a una transfusión. |
- Cinco años en archivo activo, y - Cinco años en archivo muerto |
20.4.10 |
Copias de los "Informes mensuales de la disposición de sangre y productos sanguíneos". |
Cinco años |
20.4.11 |
En su caso, registros sobre los procedimientos terapéuticos para diversos padecimientos que se hubiesen efectuado en los bancos de sangre, Centro de Colecta y servicios de transfusión. (véase el Apéndice A Normativo de este proyecto de Norma). |
Tres años |
20.5 Transcurridos los tiempos de conservación referidos en la Tabla 38 de este proyecto de Norma, el destino de los documentos y registros deberán ser destruidos o bien se conservarán en archivo muerto, bajo estricta custodia para garantizar su confidencialidad.
21 Concordancia con normas internacionales y mexicanas
Este proyecto de Norma Oficial Mexicana es parcialmente equivalente con los lineamientos y recomendaciones emitidos por Organización Mundial de la Salud, Organización Panamericana de la Salud, The European Committee on Blood Transfusion, The Association for the Advancement of Blood & Biotherapies y la International Organization for Standardization y no tiene equivalencia con Normas Mexicanas por no existir referencia al momento de su actualización.
22 Bibliografía
22.1 Farmacopea de los Estados Unidos Mexicanos, Suplementos para Dispositivos Médicos.
22.2 Standards for Blood Banks and Transfusion Services, American Association of Blood Banks. 25th edition, 2008.
22.3 Standards for Immunohematology Reference Laboratories, 5th edition, 2007. American Association of Blood Banks.
22.4 Estándares de Acreditación en Transfusión Sanguínea (2019). Asociación Española de Hematología y Hemoterapia y Sociedad Española de Transfusión Sanguínea. Rev. 2022. 5a. edición, España.
22.5 Estándares de trabajo para servicios de sangre. Organización Panamericana de la Salud. Área de Tecnología y Prestación de Servicios de Salud. Unidad de Medicamentos Esenciales, Vacunas y Tecnología. Washington, D.C., 2005.
22.6 Guidelines for national authorities on quality assurance for good manufacturing practices for biological products. WHO TRS 822 (1992).
22.7 Directrices de la OMS sobre buenas prácticas de manufactura para centros de sangre. Anexo 4 del cuadragésimo Quinto informe del Comité de Expertos de la OMS sobre especificaciones para preparados farmacéuticos. Washington, D.C.: Organización Panamericana de la Salud; 2021. Licencia: CC BY-NC-SA 3.0 IGO. https://doi.org/10.37774/9789275323366.
22.8 Guide to the preparation, use and quality assurance of blood components, 21th edition 2023. European Committee (Partial agreement) on Blood Transfusion (CD-P-TS) Recommendation No. R (95) 15.
22.9 Componentes Básicos de un Sistema Nacional de Sangre. José Ramiro Cruz. Pan American Journal of Public Health 13 (2/3):79-84 2003. Págs. 1-9.
22.10 Guía para el uso clínico de la sangre. Secretaría de Salud, Asociación Mexicana de Medicina Transfusional, A.C. y Agrupación Mexicana para el Estudio de la Hematología. México, 2007.
22.11 Terapia Transfusional en Pediatría (2009). Bravo Lindoro, A.G. Asociación Mexicana de Medicina Transfusional, A.C. Editorial Prado. México.
22.12 Gestión de Servicios de Transfusión de Sangre (1991). Editado por Hollán S.R. y Cols. Organización Mundial de la Salud. España.
22.13 Aspectos Clínicos en Medicina Transfusional (2004). Bonifaz Gracias R. y Rojo Medina J. Intersistemas, México.
22.14 El Banco de Sangre y la Medicina Transfusional (2004). Rodríguez Moyado H. Editorial Médica Panamericana. México.
22.15 Revised Classification System for HIV Infection and Expanded Surveillance Case Definition for AIDS Among Adolescents and Adults Arch Dermatol. 1993;129(3):287-290.
22.16 La cadena de frio de la sangre. Guía para la selección y adquisición de equipos y accesorios. Departamento de Tecnologías Sanitarias Esenciales. Organización Mundial de la Salud. 2004.
22.17 Manual on the management, maintenance and use of blood cold chain equipment. World Health Organization 2005.
23 Observancia de la norma
La vigilancia de la aplicación de este proyecto de Norma corresponde a la Secretaría de Salud y a los gobiernos de los estados en el ámbito de sus respectivas competencias.
24 Vigilancia de la norma
La vigilancia del cumplimiento de este proyecto de Norma corresponde a la Secretaría de Salud y a los gobiernos de las entidades federativas en el ámbito de sus respectivas competencias.
25 Vigencia
El presente proyecto de Norma entrará en vigor a los 180 días naturales posteriores cuando se lleve a cabo la publicación de la Norma Oficial Mexicana en el Diario Oficial de la Federación, toda vez que el presente documento se emite para una consulta pública durante 60 días.
TRANSITORIO
ÚNICO. La entrada en vigor del presente Proyecto de Norma, dejará sin efectos a la Norma Oficial Mexicana NOM-253-SSA1-2012 "Para la disposición de sangre humana y sus componentes con fines terapéuticos" publicada en el Diario Oficial de la Federación el 26 de octubre de 2012.
Ciudad de México, a 17 de octubre de 2024.- La Comisionada Federal para la Protección contra Riesgos Sanitarios y Presidenta del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, Armida Zúñiga Estrada.- Rúbrica.
APÉNDICE A NORMATIVO
PROCEDIMIENTOS TERAPÉUTICOS PARA DIVERSOS PADECIMIENTOS
A.1 Los procedimientos terapéuticos tales como extracciones de sangre en pacientes con poliglobulia, citaféresis reductivas o recambios plasmáticos en pacientes con leucemias, mielomas, trastornos autoinmunes y otros, podrán hacerse en bancos de sangre en coordinación con el médico tratante o directamente en los establecimientos para la atención médica especializados.
Las extracciones de sangre en pacientes con poliglobulia podrán efectuarse en bancos de sangre, Centro de Colecta o en cualquier establecimiento de atención médica.
A.2 Los procedimientos terapéuticos a que se refiere este apéndice, que se realicen en los bancos de sangre o servicios de transfusión deberán ser supervisados por el responsable sanitario. Tratándose de los Centro de Colecta por el encargado del mismo.
A.3 Tratándose de extracciones de sangre en pacientes con poliglobulia, efectuadas en bancos de sangre, Centro de Colecta, servicios de transfusión, se deberá observar lo siguiente:
a) La sangre no deberá emplearse con fines transfusionales, y
b) La bolsa contenedora se mantendrá separada del resto de las unidades y estará adecuadamente identificada, con una leyenda que diga "NO TRANSFUNDIRSE" o cualquier otra medida, a criterio del responsable sanitarios del banco de sangre o del servicio de transfusión, o bien, del encargado del Centro de Colecta, en tanto se le da destino final a la brevedad.
A.4 Para que un banco de sangre, Centro de Colecta o servicio de transfusión, efectúe algún procedimiento terapéutico deberá contar con lo siguiente:
a) La prescripción escrita del médico tratante, acompañada de los datos siguientes:
- Los de identificación del paciente y su diagnóstico, y
- Una descripción detallada del procedimiento que se requiere efectuar, incluyendo, en su caso, la recomendación de uso o contraindicación de soluciones o fármacos;
b) La carta del consentimiento informado, firmado o con la huella dactilar del paciente, recabado por el médico tratante;
c) Aprobación para llevar a cabo el procedimiento por parte del responsable sanitario del Banco de Sangre, Centro de Colecta o servicio de transfusión;
d) Equipos, materiales e insumos necesarios para la realización del procedimiento de que se trate y los necesarios para la atención de complicaciones que pudieran presentarse, incluyendo acceso a servicios de atención médica de urgencia;
e) Equipos, materiales e insumos necesarios para el destino final de los residuos biológicos que se generen, de conformidad con las demás disposiciones aplicables;
f) Personal profesional o técnico capacitado en los procedimientos, que actuará siempre bajo supervisión médica, y
g) Procedimientos normalizados de operación debidamente actualizados.
A.5 Los bancos de sangre, Centro de Colecta o servicios de transfusión, deberán conservar registros de los pacientes atendidos, los que deberán contener las acciones realizadas y sus resultados, incluyendo los volúmenes extraídos o de recambio y, en su caso, uso de soluciones o fármacos, así como efectos adversos y su manejo.
Los procedimientos y acciones realizadas deberán quedar registrados en la historia clínica del paciente.
A.6 Los bancos de sangre, Centro de Colecta o servicios de transfusión, deberán elaborar un reporte detallado al médico tratante, indicando lo referido en este apartado.
A.7 De conformidad con la Ley y el Reglamento, los establecimientos para la atención médica que utilicen equipos de aféresis automatizada para efectuar los procedimientos terapéuticos a que hace referencia este Apéndice, deberán abstenerse de realizar actos de disposición de productos sanguíneos para fines transfusionales.
APÉNDICE B NORMATIVO
PRECEPTOS DE CONTROL DE CALIDAD
B.1 Determinación de pH.
B.1.1 La determinación de pH se realiza mediante la medición de la diferencia de potencial entre un par de electrodos adecuados sumergidos en una misma solución.
B.1.2 No deberán emplearse tiras comerciales de determinación de pH para el control de calidad de productos sanguíneos.
B.1.3 El dispositivo que se utiliza para la determinación de pH es el potenciómetro
B.1.4 Los potenciómetros disponibles pueden estar equipados o no con un sistema de compensación de la temperatura.
B.1.5 Para que la calibración sea eficaz es fundamental que el electrodo se encuentre en buen estado, limpio, hidratado y sin daños físicos.
B.1.6 El electrodo debe sumergirse en la solución de almacenamiento por un tiempo aproximado de 30 minutos antes de ser utilizado.
B.1.7 Las soluciones de calibración (solución reguladora acuosa) se preparan empleando patrones primarios o reactivos analíticos de calidad apropiada.
B.1.8 Para la calibración se realiza cerca del punto isopotencial (la señal producida por un electrodo a pH 7 es 0 mV a 25ºC) y una segunda a pH 4 y/o 9.1. Se recomienda utilizar soluciones con pH cercano a 6.4 que es el punto de decisión de aceptación para los concentrados de plaquetas.
B.1.9 La calibración se puede realizar de dos maneras:
B.1.9.1 Dos puntos.
B.1.9.2 Multipunto.
B.2 Determinación de hemólisis.
B.2.1 El porcentaje de hemólisis de la masa eritrocitaria es un indicador de la calidad e integridad de los eritrocitos que permite valorar la conservación del hemocomponentes a lo largo del tiempo.
B.2.2 Se realiza con un equipo que consiste en un fotómetro y una microcubeta diseñados especialmente para la determinación cuantitativa de bajas concentraciones de hemoglobina en muestras de suero, plasma y soluciones acuosas (solución aditiva).
B.2.3 Como procedimiento alternativo para la determinación de hemólisis se puede emplear:
B.2.3.1 Método espectrofotométrico,
B.2.3.2 Método fotométrico y
B.2.3.3. Técnica en microplaca.
B.3 Determinación de proteínas totales.
B.3.1 La determinación de proteínas totales en los plasmas frescos se realizará utilizando la técnica de refractometría.
B.3.2 El resultado se reporta en gramos por litro (g/L) y se compara con el requisito de calidad exigido
B.4 Sangre Total.
B.4.1 El control de calidad de la Sangre Total (ST) comienza desde la obtención y durante toda la extracción deberá mantenerse en agitación constante, con la utilización de balanzas mezcladoras las cuales deben estar bajo control.
B.4.2 Se deberá contar con su programa de mantenimientos preventivos y los registros que demuestren que se han realizado, así como el control diario de peso y/o volumen antes de su uso para garantizar la recolección del volumen requerido.
B.4.3 Inspeccionar visualmente todas las unidades. En caso de que la unidad tenga evidencia de ruptura, daño o defecto en la bolsa, aire excesivo o cualquier otra alteración como presencia de coágulos, debe darse destino final.
B.4.4 Determinar la frecuencia de muestreo de acuerdo a la producción mensual y programar. Es conveniente realizar semanalmente el control de calidad.
B.4.5 Seleccionar aleatoriamente unidades para realizar el control de calidad, es recomendable utilizar aproximadamente 5 a 10 unidades cada semana de acuerdo a la obtención mensual, esto es, para tener una muestra representativa y valorar adecuadamente el inicio del proceso de procesamiento.
B.4.6 Como parte del control de calidad es importante calcular los pesos máximos y mínimos de la sangre total posterior a su obtención tomando en cuenta los volúmenes del límite superior e inferior permitido como requisito de calidad.
B.4.7 Estos datos deben ser colocados en el área de flebotomía y todo el personal debe cumplir con los pesos mínimos y máximos, éstos datos deben actualizarse si existe un cambio de bolsa de recolección.
B.4.8 Las unidades de sangre total de bajo volumen que se encuentren en el rango de 300 a 404 mL, aunque se pueden utilizar, éstas no se deben incluir en la estadística mensual salvo que se encuentren en un porcentaje elevado de las unidades que se estudien, sin embargo, se deberán analizar las causas e implementar acciones con la finalidad de que no ocurra con mayor frecuencia.
B.5 Concentrado eritrocitario de sangre total
B.5.1 Posterior al procesamiento:
B.5.1.1 Inspeccionar visualmente todas las unidades. En caso de que la unidad tenga evidencia de ruptura, daño o defecto en la bolsa, aire excesivo o cualquier otra alteración con la presencia de coágulos, debe darse destino final.
B.5.1.2 Determinar la frecuencia de muestreo de acuerdo a la producción mensual y programar, es conveniente realizar semanalmente el control de calidad.
B.5.1.3 Seleccionar aleatoriamente unidades para realizar el control de calidad. Es recomendable utilizar aproximadamente 5 a 10 unidades semanales de acuerdo a la obtención mensual, esto es, para tener una muestra representativa, es recomendable hacer el control de calidad de concentrados eritrocitarios que provienen de las sangres totales previamente estudiadas con la finalidad de dar seguimiento al procesamiento, principalmente la centrifugación.
B.5.1.4 Si al concentrado de eritrocitos ha sido filtrado, CE leucodepletado, la determinación de leucocitos residuales se realizará de acuerdo a la técnica descrita en el capítulo 6 de esta guía. El requisito de calidad de la cuenta de leucocitos residuales del CE leucodepletado se encuentra referenciado en la Tabla 2 parámetros de calidad de concentrados de eritrocitos.
B.5.1.5 Si se cuenta con una unidad que haya llegado a su término de vigencia determinar el porcentaje de hemólisis de la masa eritrocítica la cual no deberá ser mayor del 0.8% de acuerdo a la Guía Nacional de Control de Calidad de Sangre y Componentes Sanguíneos, disponibles en el sitio: https://www.gob.mx/cnts/documentos/recursos-guias-y-documentos-en-medicina-transfusional
B.5.1.6 Para determinar el volumen de los CE´s que contienen solución aditiva se debe determinar de acuerdo al proceso que se realice, estableciendo máximos y mínimos, de acuerdo al tipo de bolsa, utilizando herramientas estadísticas.
B.5.1.7 Si se encuentran unidades cuyo volumen se encuentre por debajo de los rangos mínimo o máximo del volumen establecido por control estadístico de proceso, las unidades NO deben ser descartadas si solo se basa en el volumen. Se debe analizar y tener un protocolo de rescate y recuperación de éstos CE´s, ajustando volumen en concordancia con otros parámetros (Hb, Hto y cuenta de leucocitos).
B.6 Plasma fresco, plasma desprovisto de factores lábiles y crioprecipitado.
B.6.1 Si se extrae ST fresca se debe anotar hora de inicio y término de la flebotomía, el tiempo no debe exceder 15 minutos. En caso de que se exceda ese tiempo se procederá a obtener el CE por centrifugación a alta velocidad y el plasma debe darle destino final por pérdida de propiedades procoagulantes.
B.6.2 Se debe anotar hora de extracción e inicio de procesamiento, si el tiempo entre la obtención y el procesamiento exceden las 6 a 8 horas, el plasma deberá ser clasificado como PLASMA DESPROVISTO DE FACTORES LÁBILES y no se considerará como plasma fresco.
B.6.3 Si el plasma es obtenido por procedimientos de aféresis (plasmaféresis) no deben exceder 6 horas posterior a su extracción y congelación para ser considerado Plasma Fresco (PF).
B.6.4 Inspeccionar visualmente todas las unidades. En caso de que la unidad tenga lipemia o una coloración no usual, evidencia de ruptura, daño o defecto en la bolsa, aire excesivo o cualquier otra alteración como exceso de glóbulos rojos, debe darse destino final.
B.6.5 Determinar la frecuencia de muestreo de acuerdo a la producción mensual y programar, es conveniente realizar semanalmente el control de calidad.
B.6.6 Seleccionar aleatoriamente unidades para realizar el control de calidad, es recomendable utilizar aproximadamente 5 a 10 unidades semanales de acuerdo a la obtención mensual, esto es, para tener una muestra representativa.
B.6.7 En caso de incumplimiento en el resultado de la celularidad, investigar las causas probables y aplicar acciones correctivas.
B.7 Crioprecipitado
B.7.1 Los crioprecipitados se descongelan a una temperatura de 37ºC y se conservan de 22 a 24ºC o en refrigeración de 2 a 6ºC.
B.7.2 Se toma una muestra para realizar la determinación de FVIIIc. Este análisis dependerá si el crioprecipitado es utilizado para transfundir pacientes hemofílicos, si no se requiere para estos pacientes no se realiza.
B.7.3 La muestra también se utiliza para determinar el Factor vW (FvW). Este análisis dependerá si el crioprecipitado es utilizado para transfundir pacientes deficientes de FvW, si no se requiere para estos pacientes no se realiza.
B.8 Determinación de Factor VIIIc.
B.8.1 Se emplea una técnica coágulométrica para medir la actividad del factor VIIIc, que es un factor de coagulación de origen proteico.
B.8.2 La muestra debe ser procesada antes de ser congelada.
B.8.3 En caso de que la muestra sea congelada se deberá realizar un protocolo de verificación para determinar en qué medida se afecta la actividad del FVIIIc.
B.8.4 Identificar correctamente la muestra en caso de que sea congelada y evitar que se borre.
B.8.5 Si la muestra se congela, las condiciones de congelación deberán ser las mismas en las que se conservan los plasmas de origen.
B.8.6 La muestra deberá permanecer en todo momento de la determinación en un recipiente de preferencia con hielo escarchado para evitar degradación del FVIIIc.
B.8.7 En el caso de que la muestra sea maquilada, se deberá elaborar un protocolo de conservación y transporte asegurando que el factor VIIIc no se deteriore durante el transporte al sitio donde se hará la determinación.
B.8.8 Si la muestra fue congelada, debe ser descongelada a temperatura controlada de 37ºC en baño maría y posteriormente conservada a una temperatura de 20 -24ºC, preferentemente si se cuenta con el recurso en un recipiente con hielo escarchado.
B.8.9 El tiempo utilizado durante la realización de la prueba es crucial por lo que no debe tardar más del tiempo necesario, debido a que puede haber degradación del FVIIIc, este tiempo debe ser analizado y establecido por el laboratorio que realice la prueba con base a sus procedimientos.
B.8.10 Como parte del control de calidad de la prueba se recomienda procesar como testigo una mezcla de plasma humano normal.
B.8.10.1 Cada vez que se realiza la prueba se descongela un tubo de plasma normal como testigo y se trabaja a la par de la muestra problema, de esta manera se busca tener controlada la prueba.
B.9 Determinación de Fibrinógeno.
B.9.1 Esta determinación se puede realizar con cualquier instrumento que haga la prueba de Tiempo de trombina y dependerá de las instrucciones del fabricante.
B.9.2 Debido a que el resultado que se obtiene es en medida de tiempo (segundos) se debe utilizar un calibrador con una concentración conocida de fibrinógeno en mg/mL para elaborar la curva de calibración.
B.10 Determinación de Factor de von Willebrand (FvW).
B.10.1 Existen diferentes técnicas para determinar el FvW, la más comúnmente utilizada es llamada "Actividad del factor de von Willebrand-cofactor de ristocetina", que permite evaluar el funcionamiento de la proteína de vW, que interviene en la coagulación de la sangre.
B.10.2 Aunque no todos los laboratorios cuentan con la prueba en ocasiones se podrá enviar las muestras a maquilar, por lo que, es importante realizar un procedimiento validado de transporte de la muestra al laboratorio.
B.10.3 Esta técnica solo se deberá realizar cuando el crioprecipitado sea utilizado en pacientes con Enfermedad de von Willebrand, para verificar la calidad del componente que será transfundido.
B.11 Concentrado Plaquetario.
B.11.1 El control de calidad de los concentrados plaquetarios debe realizarse semanalmente y contar con un programa de muestreo haciendo un seguimiento desde la unidad de ST de la cual se obtuvo la capa leucoplaquetaria, para verificar y asegurar todas las condiciones de procesamiento, centrifugación y obtención, así como después de cada mantenimiento preventivo o correctivo de los equipos involucrados en el proceso general.
B.11.2 La frecuencia del muestreo también se puede determinar por el control estadístico de proceso.
B.11.3 Inspeccionar visualmente todas las unidades. En caso de que la unidad tenga evidencia de ruptura, daño o defecto en la bolsa, aire excesivo o cualquier otra alteración como agregados plaquetarios, debe darse destino final.
B.11.4 Todos los concentrados plaquetarios deben ser inspeccionados visualmente, en cada etapa del procesamiento e inmediatamente antes de ser distribuidos a los servicios de transfusión u otros bancos de sangre.
B.11.5 La inspección visual deberá realizarse en un espacio con las condiciones de iluminación adecuadas, verificando lo siguiente:
B.11.5.1 Presencia de "remolino".
B.11.5.2 Ausencia de agregados plaquetarios o restos de fibrina.
B.11.5.3 Ausencia de coágulos.
B.11.5.4 Los concentrados plaquetarios que tengan contaminación eritrocitaria mayor a 1 mL, pueden utilizarse para uso transfusional, realizando las pruebas pretransfusionales correspondientes.
B.11.5.5 Las unidades con coloración verde (secundaria al uso de anticonceptivos orales) o naranja (secundaria al consumo de vitamina A y carotenos), son aceptables para su uso transfusional.
B.11.6 Determinar la frecuencia de muestreo de acuerdo con la producción mensual y programar, es conveniente realizar semanalmente el control de calidad o determinar la frecuencia por medio del control estadístico de proceso.
B.11.7 Seleccionar aleatoriamente unidades para realizar el control de calidad, es recomendable utilizar aproximadamente 5 a 10 unidades semanales de acuerdo con la obtención mensual, esto es, para tener una muestra representativa es recomendable hacer el CC de los CPs que provienen de las ST´s previamente estudiadas con la finalidad de dar seguimiento al procesamiento, principalmente la centrifugación.
B.11.8 Para el caso de las aféresis o mezcla de concentrados plaquetarios, para poder realizar el control bacteriológico es conveniente utilizar un conector estéril y transferir la cantidad necesaria a una bolsa de transferencia y cultivar inmediatamente para evitar contaminación por mal manejo o realizar el cultivo en condiciones de esterilidad y retornar el componente al inventario.
B.11.9 Las determinaciones analíticas se deberán realizar preferentemente después de las 24 horas posteriores a la obtención del CP, tanto de aféresis como los unitarios y las mezclas, debido a que posterior al procesamiento las plaquetas tienden a formar grumos los cuales se disuelven por efecto de la agitación.
B.11.10 Conservar la unidad a una temperatura de cuarto entre 20-24ºC, y mantenerla en agitación constante para evitar que las plaquetas se agreguen, es conveniente colocar la unidad de manera que la etiqueta no obstruya el intercambio gaseoso de la unidad.
B.11.11 Si los concentrados plaquetarios fueron sometidos a procesos de filtración prealmacenamiento, es necesario recolectar una muestra antes de comenzar el proceso de filtrado y otra al concluir.
B.11.12 En caso de incumplimiento en el resultado de la celularidad, investigar las causas probables y aplicar acciones correctivas.
B.11.13 La medición del pH se debe realizar al término de la vigencia debido a que es un indicador de la conservación y almacenamiento de la unidad, y su disminución se puede atribuir a las lesiones que sufren las plaquetas por el almacenamiento, adicionalmente es un método indirecto que permite detectar contaminación bacteriana y evaluar lesiones ocurridas en las plaquetas durante su almacenamiento.
B.12 Leucocitos Residuales en Procesos de Leucodepleción.
B.12.1 Las muestras para conteo de leucocitos residuales deben ser obtenidas y evaluadas dentro de las primeras 72 horas posteriores a la donación, debido a que los leucocitos sufren deterioro al mantenerse refrigerados. Cuando el componente a estudiar contenga glóbulos rojos, se recomienda que el hematocrito no exceda 60%.
B.12.2 Se puede realizar recuento en Cámara de Nageotte de leucocitos residuales en productos sanguíneos leucodepletados o bien por citometría de flujo.
B.13 Control de calidad y verificación del proceso de asepsia.
B.13.1 El control de calidad y verificación del proceso de asepsia se puede realizar por
B.13.1.1 Método por placas de contacto. El uso de placas de contacto de 65 mm de diámetro que contienen agentes para neutralización se ha descrito como un método para evaluar la eficacia del proceso de desinfección. Consiste en realizar cultivos de las placas después de ponerlas en contacto durante 30-40 segundos con la piel de la fosa anticubital del brazo de los donantes antes y después de la desinfección. Posteriormente, las placas se colocan en una incubadora a 37 °C durante 24-48 horas para contar las colonias presentes.
B.13.1.2 Métodos de cultivo tradicional. Incluyen el muestreo con un hisopo estéril, siembras de cultivos primarios, con incubaciones de 2 hasta 7 días, seguidos de un cultivo secundario en placas incubadas por 2 días más, para finalmente realizar de manera adicional la identificación por pruebas bioquímica. Estas técnicas por cultivo tradicional representan una alternativa para el método de placas por contacto.
B.13.1.3 La metodología para la obtención y siembra de muestras de productos sanguíneos para cultivo microbiológico pueden encontrarse en el apartado G. Anexo 1 de la Guía de control de calidad microbiológico de componentes plaquetarios para incrementar la seguridad en su uso clínico en México.
B.14 Control de calidad de equipos.
Los requisitos mínimos para el control de otros equipos se muestran en la abla B.14 de esta Norma
Tabla B.14. Requisitos mínimos para el control de equipos de uso común en laboratorio de banco de sangre
Equipo |
Forma de verificación |
Periodicidad de la verificación |
Frecuencia de calibración o equivalente |
Báscula pesa personas con altímetro |
Estandarizar con patrón de peso calibrado y con regla graduada |
Semestral |
Anual o cada vez que sea necesario. |
Termómetro de laboratorio |
Comparar con termómetro patrón calibrado. |
A su estreno |
Anual o cuando sea necesario. |
Refrigeradores, congeladores, cámaras frías y equipos para conservación de plaquetas |
Comparar con termómetro patrón calibrado. |
Semestral |
Mantenimiento anual o cada vez que sea necesario. |
Indicador de temperatura |
Comparar con termómetro de laboratorio. |
Cada día de uso |
Mensual y cuando sea necesario. |
Reloj de laboratorio |
Comparar con cronómetro calibrado. |
Mensualmente |
Anual o cuando sea necesario. |
Gabinete de seguridad biológica clase II, y Campana de flujo laminar |
Condiciones de limpieza. |
Diariamente |
En su caso tomar acciones correctivas. |
Control bacteriológico. |
Mensualmente |
Anual o cuando sea necesario. |
|
Control de medición de micropartículas |
|||
Centrífuga refrigerada |
Observar los indicadores de velocidad y temperatura. |
Cada día de uso |
Anual o cuando sea necesario; el servicio de mantenimiento técnico hará control de precisión de revoluciones por minuto, aceleración y frenado. |
Revoluciones por minuto, medidas por medio de fototacómetro calibrado. |
Cada seis meses |
||
Temperatura, comparando con termómetro patrón calibrado. |
Cada seis meses |
||
Comprobar que los productos obtenidos, reúnan los requisitos de calidad establecidos. |
Mensualmente |
||
Centrífuga de mesa para laboratorio clínico y centrífuga de mesa para pruebas serológicas |
Verificar la separación adecuada de los compuestos de diferente densidad o de las partículas de diferente tamaño suspendidas en un líquido. |
Cada día de uso |
Anual o cuando sea necesario. |
Verificar las revoluciones por minuto, empleando un fototacómetro calibrado. |
Cada seis meses |
||
Centrífuga para hematocrito |
Comprobar la ausencia de hemólisis y que la capa leucoplaquetaria e interfase plasma-células estén bien definidas. |
Cada día de uso |
Anual o cada vez que sea necesario. |
Revoluciones por minuto, empleando un fototacómetro calibrado. |
Cada seis meses |
||
Tipificador sanguíneo automatizado |
Hacer controles comparativos. |
Cada día de uso |
Anual o cuando sea necesario. |
Fotómetro (para medición de hemoglobina) |
Utilizar el control del fabricante. |
Cada día de uso |
Anual o cuando sea necesario. |
Contadores celulares |
Utilizando control del fabricante. |
Cada día de uso |
Anual o cuando sea necesario. |
Baño María y bloques térmicos |
Verificar temperatura con termómetropatrón calibrado. |
Cada día de uso |
Anual o cuando sea necesario. |
Micropipetas y dispensador automático |
Verificar la exactitud del volumen mediante balanza analítica. |
Cada cuatro meses |
Anual o cuando sea necesario. |
Autoclave |
Comprobar efectividad con indicadores biológicos. |
Cada vez que se utilice |
Anual o cuando sea necesario. |
Agitadores serológicos |
Observar rotación y panel de controles. |
Cada día de uso |
Cada seis meses y ajuste de velocidad cada vez que sea necesario. |
Báscula para bolsas de recolección |
Estandarizar con patrón de peso calibrado. |
Semestral |
Cada seis meses o cada vez que sea necesario. |
Mezclador de sangre automatizado con control de volumen |
Verificar el peso de la primera bolsa de sangre recolectada mediante balanza verificada. |
Cada día de uso |
Cada seis meses o cada vez que sea necesario. |
Potenciómetro |
Empleando soluciones amortiguadoras de pH bajo y alto. |
Cada vez que se hagan mediciones |
Anual o cuando sea necesario. |
Sistemas de conexión estéril |
Comprobar sellado adecuado. |
Cada día de uso |
Cuando sea necesario. |
Sensor de temperatura y sistema de alarma de calentadores para sangre y componentes sanguíneos |
Comparar temperatura con termómetros de laboratorio. |
Cada seis meses |
Anual o cuando sea necesario. |
APÉNDICE C NORMATIVO
EMBALAJE Y TRANSPORTE PARA CONSERVACIÓN DE SANGRE Y PRODUCTOS SANGUÍNEOS
C.1 Contenedores para embalaje de sangre y productos sanguíneos.
C.1.1 Podrán ser utilizados como embalaje externo para la conservación y transporte de bolsas sangre y productos sanguíneos; en orden de preferencia se pueden utilizar los siguientes dispositivos, cumpliendo con las especificaciones mínimas señaladas:
C.1.1.1 Caja isotérmica para el transporte y conservación de 1 hasta 24 horas de bolsas de sangre y productos sanguíneos: elaborada en material resistente, durable y anticorrosivo, cierre hermético, conformada por paredes dobles inyectadas con espuma para un mejor aislamiento térmico, resistente a caídas y apilamiento. La capacidad será determinada de acuerdo a las necesidades específicas, considerando el espacio para los elementos de enfriamiento eutéctico (-32 °C, +4 °C, +22 °C, +37 °C) y las bolsas de sangre y/o productos sanguíneos.
C.1.1.2 Nevera o termo isotérmico portátil: de plástico sólido, adecuado a la capacidad y el peso de los hemocomponentes a transportar. De fácil limpieza y desinfección con los productos habituales usados para este fin. Debe asegurar el cierre hermético para evitar que apertura durante el transporte, así como garantizar la cadena de conservación y evitar la contaminación del contenido. Para la protección de los hemocomponentes se podrá utilizar un material amortiguador adecuado (papel absorbente, plástico burbuja, entre otros). Será empleada únicamente para transporte y conservación que no conlleve más de 4 horas.
C.1.1.3 Nevera o termo portátil de poliestireno: de tamaño adecuado a la capacidad y el peso de los hemocomponentes a transportar. Debe permitir la limpieza y desinfección con los productos habituales usados para este fin. Debe garantizar la cadena de conservación y evitar la contaminación del contenido. Para la protección de los hemocomponentes se podrá utilizar un material amortiguador adecuado (papel absorbente, plástico burbuja, entre otros). Solo será empleada para transporte y conservación que no conlleve más de 120 minutos.
C.1.2 Las muestras para el procesamiento de pruebas en bancos de sangre y los hemocomponentes sin resultados de pruebas infecciosas son catalogadas como sustancias infecciosas de categoría B, por lo que su embalaje y transporte debe realizarse de acuerdo a lo establecido para dicha categoría, por lo que no deben transportarse en el mismo contenedor (caja, nevera o termo) de productos sanguíneos liberados para uso terapéutico.
C.2 El embalaje para conservación y transporte, de cualquier tipo, deberá tener en la parte externa la siguiente información:
C.2.1 Etiqueta de identificación del tipo y material biológico que se transporta y este contenido en la caja, nevera o termo.
C.2.2 Etiqueta de estiba hacia arriba de dimensiones mínimas 74 × 105 mm, en lados opuestos. Color: blanco y negro o blanco y rojo (flechas).
C.2.3 Nombre, dirección y teléfono móvil del remitente.
C.2.4 Nombre, dirección y teléfono móvil del destinatario.
C.2.5 Condiciones de almacenamiento y temperatura de conservación del contenido.
C.2.6 Instrucciones de limpieza e inactivación del contenido, con solución de hipoclorito de sodio, en caso de derrame o rotura de los productos sanguíneos.
C.3 Cuando se use hielo seco para mantener en óptimas condiciones algunos hemocomponentes, éste deberá colocarse fuera de los embalajes secundarios, entre éste y el embalaje exterior o terciario o en un sobre embalaje; adicionalmente se colocarán cuñas interiores para que los embalajes secundarios se mantengan en su posición inicial cuando el hielo seco se haya evaporado. Cuando se utiliza dióxido de carbono sólido (hielo seco), el embalaje/envase estará diseñado y construido para que permita la salida del dióxido de carbono gaseoso y prevenir así una acumulación de presión que pudiera romper los envases, y el embalaje (embalaje exterior o sobre embalaje) deberá marcarse con la indicación «Dióxido de carbono sólido» o «Hielo seco», conforme a las instrucciones de embalaje 954 de la International Air Transport Association (IATA).
C.4 La caja transportadora, nevera o termo deberá acompañarse con una carta/oficio en hoja membretada, emitida por el servicio de sangre remitente, al servicio de sangre destinatario, señalando el número de unidades remitidas, su tipo ABO y Rh(D), la temperatura de conservación, el número de unidad, el nombre del responsable del embalaje, el responsable de autorización del envío, la dirección completa del remitente y del emisor y la leyenda de que el material biológico ha sido estudiado para infecciones transmisibles por transfusión, con resultados no reactivos. Se debe señalar que las unidades son transportadas con fines transfusionales y que carecen de valor comercial alguno
C.4.1 El Banco de Sangre, Centro de Procesamiento de Sangre, Centro de Colecta de Sangre y sus Componentes, Centro de Distribución de Sangre y Productos Sanguíneos y los Servicios de Transfusión, deben contar con un procedimiento normalizado de operación para evitar errores en la correcta identificación del termo, hielera o contenedor calificado. Adicionalmente el contenedor deberá contar con una etiqueta con la siguiente información:
C.4.1.1 Nombre del establecimiento proveniente.
C.4.1.2 Nombre del establecimiento destino.
C.4.1.3 Cantidad y nombre de las unidades colocadas en el termo o hielera o contenedor calificado.
C.4.1.4 Si o no se colocó termómetro o dispositivo calibrado para la medición de temperatura y así permitir la trazabilidad de la misma.
C.4.1.5 El rango de temperatura en que deben conservarse y transportar, se señalará con números y signos, por ejemplo: (+ 2°C a + 10 °C, + 20°C a + 24°C, - 20°C a - 25°C).
C.4.1.6 El tiempo máximo de transporte que garantice el rango de temperatura.
C.4.1.7 Se le anotará la leyenda: "NO ABRIR", "NO ABRIR HASTA SU DESTINO" o cualquier otra medida que garantice la conservación óptima.
C.4.1.8 Identificación numérica o alfanumérica exclusiva para cada envío del termo o hielera o contenedor calificado.
C.4.1.9 Firma o iniciales de quién acondicionó y quién validó el armado o acondicionamiento del termo o hielera o contenedor calificado.
C.5 Protocolo de calificación de contenedores de sangre.
C.5.1 Todos los contenedores empleados para transporte de sangre y productos sanguíneos deberán estar calificados para la conservación de temperatura conforme al componente sanguíneo a transportar tomando como referencia el concepto de autonomía frigorífica , definiendo en cada caso, el embalaje adecuado, la cantidad y tipo de refrigerante empleado para cada uno de los eventos de transporte, considerando los tiempos de traslado estimados entre el servicio de sangre remitente y el servicio receptor, siendo obligación del responsable sanitario verificar que el protocolo de acondicionamiento para el transporte se cumple adecuadamente.
C.5.2 Una operación de calificación requiere un número suficiente de monitores de registro de datos de temperatura electrónicos de acuerdo al tamaño de la instalación, para garantizar que las actividades de calificación se puedan llevar a cabo correctamente. Además, se necesitan equipos informáticos para almacenar y analizar los datos. La calificación debe contar con las siguientes etapas:
C.5.2.1 Debe contar con calificación de diseño (CD) basada en los requerimientos de usuario.
C.5.2.2 Debe contar con calificación de instalación (CI) con base en los requisitos del fabricante.
C.5.2.3 Debe contar con calificación de operación (CO) basada en las condiciones e intervalos de operación establecidas por el fabricante y usuario.
C.5.2.4 Debe contar con calificación de desempeño (CE) que demuestre que el equipo y sistema cumple con los requisitos previamente establecidos en condiciones de uso rutinario y dentro de los intervalos de trabajo permitidos para cada producto.
C.5.2.5 No se podrá continuar con la siguiente etapa de calificación, sin antes haber concluido satisfactoriamente la precedente.
C.5.2.6 Los instrumentos críticos de medición involucrados en la calificación, deben estar calibrados con trazabilidad a los patrones nacionales.
C.5.3 Los monitores de registro de datos de temperatura electrónicos deben cumplir con las siguientes características para ser empleados para calificar los contenedores a emplear por el servicio de sangre:
C.5.3.1 Ser técnicamente adecuados para la tarea específica y el entorno operativo previsto;
C.5.3.2 Proporcionar un registro continuo y confiable de los datos de temperatura en el tiempo;
C.5.3.3 Tener un rango de temperatura, y humedad si corresponde, adecuado para poder registrar todos los extremos de temperatura previstos (por ejemplo, de -30ºC a +60° C);
C.5.3.4 Tener un período de muestreo de datos programable por el usuario que permita establecer intervalos de tiempo en un rango de 1 a 15 minutos o más y con suficiente memoria para la duración prevista del estudio y el intervalo de registro elegido;
C.5.3.5 Tener un certificado de calibración en 3 puntos, con trazabilidad al Centro Nacional de Metrología (CENAM), y tener un error de no más de +/- 0.5°C en cada punto de calibración;
C.5.3.6 Permitir que los datos registrados de temperatura y tiempo se descarguen en un sistema informático para su posterior análisis;
C.5.3.7 Tener un software analítico y de almacenamiento de datos.
C.5.4 Los Servicios de Sangre deben contar con un Plan Maestro de Validación o un equivalente escrito para el desarrollo de las actividades de calificación y validación, el cual debe ser un documento conciso, claro y autorizado por el mayor nivel jerárquico de la organización (responsable sanitario y responsable de calidad)), en donde debe quedar establecido el alcance, las responsabilidades y las prioridades de la calificación y validación, que incluya al menos:
C.5.3.1 Política de validación y calificación
C.5.3.2 Vigencia
C.5.3.3 Alcance
C.5.3.4 Objetivos generales y específicos.
C.5.3.5 Estructura organizacional para las actividades de validación
C.5.3.6 Descripción de las instalaciones, equipos, sistemas, métodos y procesos a calificar y/o validar.
C.5.3.7 Formatos a emplearse para los protocolos y reportes.
C.5.3.8 Planeación y programación de las calificaciones/validaciones.
C.5.3.9 Control de cambios.
C.5.3.10 Referencia a documentos aplicables.
C.5.3.11 Métodos analíticos.
C.5.3.12 Programas o sistemas computacionales que impactan a la calidad del producto.
C.5.3.13 Sistemas críticos.
C.5.3.14 Equipo de procesamiento y acondicionamiento.
C.5.3.15 Procedimiento o métodos de limpieza y/o sanitización.
C.5.3.16 Procesos de procesamiento y acondicionamiento.
C.5.3.17 Programa de actividades, el cual deberá ser actualizado con la frecuencia requerida.
C.5.3.18 Mantenimiento del estado validado.
C.6 Vehículos de transporte para unidades de sangre y sus componentes de Centros de Distribución de Sangre.:
C.6.1 Los servicios de sangre de los sectores público, social y privado, deberán vigilar el funcionamiento adecuado de la red o cadena de frío, disponiendo para ello de equipo y personal capacitado en los Procedimientos Normalizados de Operación de almacenamiento, conservación, distribución, control y transporte.
C.6.2 Es responsabilidad del establecimiento que envía las unidades garantizar que los vehículos y equipos utilizados para distribución sean adecuados para su uso y estén equipados adecuadamente para prevenir la exposición de las unidades a condiciones que podrían afectar su calidad e integridad.
C.6.3 Contar con la calificación del transporte y calibración vigente de los instrumentos que se encuentran en el transporte que permitan monitorear y verificar la trazabilidad de la temperatura durante el traslado para asegurar la conservación óptima. Estos deberán estar sujetos a programas de mantenimiento periódico y los instrumentos de medición deberán tener fechas de calibración vigentes.
C.6.4 Deben existir procedimientos normalizados de operación para la operación y mantenimiento de todos los vehículos y equipos utilizados para el proceso de envío, incluyendo las precauciones de limpieza y de seguridad.
C.6.5 Siempre que sea posible deben utilizarse vehículos y equipos dedicados exclusivamente a este envío. Cuando se utilicen vehículos y equipo no dedicados se deberá contar con procedimientos para asegurar que la calidad e integridad de las unidades no se vea comprometida, estableciendo los controles que deben cumplir y las características que deben tener.
C.6.6 El Servicio de Sangre debe de proporcionar al personal designado una capacitación inicial y continua sobre la cadena de frío, basada en procedimientos normalizados de operación y de acuerdo con un programa de capacitación documentado.
C.6.7 Para el traslado de unidades de sangre y productos sanguíneos de un establecimiento a otro en transporte refrigerado, será aplicativo lo que se indica a continuación:
C.6.7.1 Se debe vigilar la temperatura, la limpieza y la seguridad de las instalaciones cuando la ruta de transporte incluye descarga y carga. Está expresamente prohibido almacenar alimentos y bebidas o cualquier otro producto ajeno a las unidades.
C.6.7.2 Es responsabilidad del transportista proteger las unidades contra la rotura, la adulteración, robo y garantizar que las condiciones de temperatura se mantengan dentro de límites aceptables durante el transporte.
C.6.7.3 El transporte se deberá realizar empleando medios refrigerantes que mantengan la temperatura que se requiera dependiendo de la unidad que se trate y tener un registrador de temperatura que permita contar con datos continuos de temperatura durante todo el transporte.
C.6.7.4 Si se utilizan vehículos con control de temperatura, el equipo de monitoreo de temperatura utilizada durante el transporte debe someterse a mantenimiento y calibrado a intervalos regulares. Debe llevarse a cabo mapeos de temperatura en condiciones representativas y debe tener en cuenta las variaciones estacionales.
C.6.7.5 Si se utilizan refrigerantes en cajas aisladas, éstas deberán ser colocadas de tal manera que las unidades no entran en contacto directo con el refrigerante. El personal debe estar capacitado en los procedimientos de embalaje de las cajas aislantes y en la reutilización de los refrigerantes.
C.6.7.6 Debe haber un sistema para controlar la reutilización de los refrigerantes para garantizar que no se usan por error refrigerantes no fríos. Debe haber una separación física adecuada entre los paquetes congelados y fríos.
C.6.7.7 Deberán estar descritos en un procedimiento el proceso de entrega de las unidades y el control de las variaciones de temperatura estacionales.
C.6.7.8 Si se produce una desviación de la temperatura o un daño a las unidades durante el transporte, se deberá informar al establecimiento y destinatario de las unidades afectadas. Se debe contar con un procedimiento para la investigación y el manejo de variaciones de temperatura.
C.6.7.9 Se deberán documentar e investigar las desviaciones a los procedimientos documentados.
C.6.7.10 Se deberán tomar las acciones correctivas y preventivas adecuadas (CAPA) para corregir las desviaciones y prevenirlas de acuerdo con los principios de la gestión de riesgos de calidad.
C.6.7.11 Tomar las medidas necesarias con la finalidad de cumplir con la validación y que las unidades durante el traslado se mantengan dentro del rango de temperatura que se indican a continuación:
C.6.7.12 De +2°C a +10°C para sangre total o reconstituida, concentrado de eritrocitos y concentrados de eritrocitos descongelados y resuspendidos y plasmas o crioprecipitados en estado líquido;
C.6.7.13 De +20°C a +24°C para preparados con plaquetas.
C.6.7.14 A temperaturas que garanticen el estado de congelación (-20° a -25°C) para los plasmas, crioprecipitados, mezclas de crioprecipitados, plaquetas o concentrados de eritrocitos congelados;
C.6.7.15 De +20°C a +24°C para unidades de granulocitos;
C.6.7.16 El servicio de sangre deberá contar con un Plan de contingencia en casa de falla del transporte.
C.6.8 En caso de utilizar un transportista externo se debe de considerar lo siguiente:
C.6.8.1 Validar y verificar si cuenta con todas las capacitaciones correspondientes para un traslado adecuado.
C.6.8.2 Cualquier actividad que se subcontrate y que cubra actividades deben estar definidas, acordadas y controladas correctamente con el fin de evitar malentendidos que puedan afectar la integridad de las unidades. Tiene que haber un contrato escrito entre el contratante y el contratista que establezca claramente las obligaciones de cada parte.
____________________________